Cell Imaging
Summary of the reagents you can use for imaging studies in virtually any field of investigation
Introduction
Researchers are increasingly adding imaging analyses to their repertoire of experimental methods for understanding the structure and function of biological systems. New methods and instrumentation for imaging have improved resolution, signal detection, data collection and manipulation for virtually every sample type. Live-cell and in vivo imaging have benefited from the availability of reagents such as vital dyes that have minimal toxicity, the discovery of intrinsically fluorescent proteins (IFPs; Stephens and Allan, 2003; Sullivan and Kay, 1999) and the development of imaging systems that do not damage biological samples (Stephens and Allan, 2003). Techniques such as fluorescent analog chemistry have allowed scientists to study the cytoskeleton in living cells, follow the recycling of cell-surface components, and look at movement and distribution of cellular proteins (Wang, 1989). Additionally, techniques for culturing cells on the microscope stage and maintaining a constant focal plane continue to improve (Stephens and Allan, 2003; McKenna and Wang, 1989) so that cellular processes can be followed in real time. New technologies such as magnetic resonance imaging have allowed in vivo imaging of processes, such as brain activity, in whole organisms (Check, 2005).
Here we present a summary of reagents that researchers can use for imaging studies in virtually any field of investigation. HaloTag® technology allows researchers to image live cells, perform multicolor imaging experiments and immunocytochemistry or potentially immunohistochemistry. We also present the Monster Green® Fluorescent Protein, an engineered intrinsically fluorescent protein (IFP) with improved signal and expression for imaging studies in mammalian cells. Additionally, we describe antibody reagents that label protein markers of specific cell types, such as the Anti-βIII Tubulin mAb. For researchers dissecting apoptosis, imaging reagents for multiple points in the pathway are also discussed, including a phosphorylation-specific antibody to caspase-3, an antibody against the p85-fragment of PARP and an anti-cytochrome c. Finally, we present phosphorylation-specific antibodies for studying a variety of cell-signaling pathways.
HaloTag® Technology
Overview
HaloTag® Technology provides new options for rapid, site-specific labeling of HaloTag® fusion proteins in living cells and in vitro. The technology is based on the formation of a covalent bond between the HaloTag® protein and synthetic ligands that carry a variety of functionalities, including fluorescent labels, affinity tags and attachments to solid supports. The covalent bond forms rapidly under physiological conditions, is highly specific and essentially irreversible, yielding a complex that is stable even under denaturing conditions. Therefore, using HaloTag requires only a single fusion construct that can be expressed, labeled with any of a variety of fluorescent moeties and imaged in live or fixed cells.

Components of the HaloTag® Technology
The HaloTag® protein is a genetically engineered derivative of a hydrolase that efficiently forms a covalent bond with the HaloTag® Ligands (Figure 1). This 34kDa monomeric protein can be used to generate N- or C-terminal fusions that can be expressed in a variety of cell types. Since the HaloTag® protein is rare and of prokaryotic origin, endogenous activity is absent from E. coli, plant and mammalian cells. This technology has been used successfully in many systems ranging from plant to human stem cells (Eisenstein, 2006; Zhang, 2006; Lang, 2006; and Shinohara and Matsubayahshi, 2007).
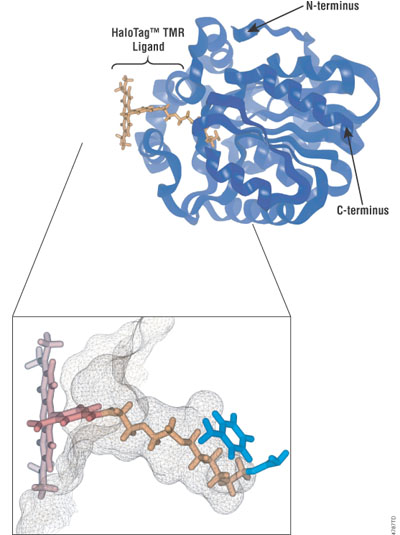
Figure 1. Molecular model of the HaloTag® protein with a covalently bound HaloTag® TMR Ligand. Overview of the protein structure (top right) with close-up of the ligand tunnel outlined by a mesh Connolly surface (lower left). The HaloTag® TMR Ligand (fluorescent moiety in red, reactive linker in orange) is shown covalently bound to the aspartate nucleophile (shown in blue). Replacement of the catalytic base (histidine) with a phenylalanine (also shown in blue) makes the protein incapable of hydrolyzing the formed covalent bond, leading to the formation of a stable covalent bond.

Figure 2. Overview of protocol for live-cell and fixed-cell imaging using the HaloTag® Technology. This technology allows labeling and analysis of HaloTag® fusion proteins by live- or fixed-cell imaging, gel-based fluoroimaging, Western blotting and capture of HaloTag® fusion proteins for proteomics applications. Note that, although this technology provides multiple alternatives to the use of antibodies, the Anti-HaloTag® pAb adds further flexibility by enabling immunocytochemistry and Western blot analysis.
A variety of HaloTag® Flexi® Vectors for mammalian, E. coli and cell-free protein expression are available. These vectors are compatible with the Flexi® Vector System, a directional cloning method for protein-coding sequences. The Flexi® Vector System provides a rapid, efficient and high-fidelity way to transfer these sequences between a variety of Flexi® Vectors. These vectors contain various expression or peptide tag options to enable expression of native or fusion proteins for studies of protein structure and function or protein:protein or protein:DNA interactions.

Figure 3. The HaloTag® 7 Flexi® Mammalian Vectors. This series of vectors for creating N-terminal or C-terminal HaloTag® fusion proteins gives researchers the ability to choose a vector with the ideal expression levels for their experimental needs. The vectors are available with either Kanamycin or Ampicillin resistance. For more details see the Promega Notes article.
We developed the HaloTag® 7 Flexi® Vector CMV deletion series, a series of human CMV promoter deletions that offer different constitutive expression levels for HaloTag® fusion proteins (Figure 2). This allows researchers to express just enough protein for their application. The vectors are available with kanamycin or ampicillin resistance markers, and vectors are available for generating either N-terminal or C-terminal fusions to the HaloTag® 7 protein. For more information on HaloTag® Flexi® Vectors see the article How to get started with the HaloTag Technology.
HaloTag® Ligands are small chemical tags that are capable of covalently binding the HaloTag® protein. These ligands contain two crucial components: 1) a common HaloTag® Reactive Linker that ensures formation of a covalent bond with the HaloTag® protein, and 2) a functional group: fluorescent or affinity tag. The HaloTag® Ligands for imaging contain fluorophores that span the visible spectrum and include cell-permeant and impermeant options.
Live-Cell Imaging Using HaloTag® Technology
With HaloTag® technology, researchers clone their gene of interest into a HaloTag® Flexi® vector. When expressed, this fusion protein can be labeled specifically and efficiently with a variety of spectrally distinct fluorescent tags. These small tags, called HaloTag® Ligands, are comprised of a linker that covalently binds to the HaloTag® protein and a fluorescent moiety. Importantly the HaloTag® protein and these ligands are completely nontoxic to cells. The HaloTag® protein itself has no endogenous eukaryotic equivalent and does not interfere with the proper cellular functioning of fusion partners.

Figure 4. Live-cell labeling with the HaloTag® Technology. Schematics show live-cell labeling options. Panel A. Rapid labeling. Panel B. No-Wash labeling.
The HaloTag® labeling technology for imaging offers a quick and simple way to label expressed HaloTag® fusions within live cells. Using this strategy, termed “Rapid” Labeling, the HaloTag® fusion can be labeled with any of a variety of cell-permeant ligands and/or an impermeant one. During the recommended short incubation in the presence of ligand, cell-permeant ligands freely enter cells and their subcellular compartments, covalently attaching to the HaloTag® fusion protein (Figure 4). A subsequent wash step allows the unbound ligand to exit cells, resulting in a highly specific signal with very low background noise (Figure 4).
Live-Cell Labeling (Rapid Protocol)
This protocol is intended for labeling live cells with the cell-permeant HaloTag® TMR, diAcFAM, Oregon Green® or Coumarin Ligands, or the cell-impermeant Alexa Fluor® 488 Ligand.
Materials Required
- HaloTag® pHT2 Vector or HaloTag® Flexi® Vectors, desired ligands and protocol #TM260
- chambered cover glass with cells expressing HaloTag® fusion protein
- complete culture medium appropriate for your cells at 37°C
- culture medium lacking phenol red at 37°C (optional)
- 1X PBS (pH 7.5, optional for washes)
- confocal microscope or wide-field fluorescent microscope equipped with appropriate filter sets and lasers
- 37°C + CO2 cell culture incubator
Rapid Labeling Procedure
- Prepare a 1:200 dilution of HaloTag® TMR, diAcFAM, Oregon Green®, Coumarin or Alexa Fluor® 488 Ligand in warm culture medium just prior to addition to cells. This is a 5X working stock solution.
- Label cells by replacing one-fifth of the existing volume of medium with the 5X HaloTag® ligand working stock solution, and mix gently. This results in the recommended final labeling concentrations of 5μM TMR; 1μM diAcFAM, Oregon Green® or Alexa Fluor® 488; and 10μM Coumarin.
- Incubate for 15 minutes in a 37°C + CO2 cell culture incubator.
- Gently replace the ligand-containing medium with an equal (or greater) volume of warm fresh medium (or 1X PBS [pH 7.5]). Repeat this two times ending with warm complete medium, for a total of three complete rinses.
- Incubate cells in complete culture medium at 37°C + CO2 in a cell culture incubator for 30 minutes to wash out unbound ligand (TMR and Alexa Fluor® 488 ligands may need only 15 minutes).
- Replace the medium with an equal volume of fresh warm culture medium (use of medium lacking phenol red may improve imaging).
- Transfer to a microscope and capture images.
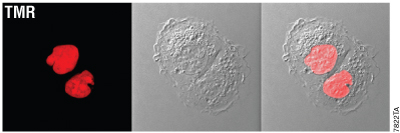
Figure 5. HaloTag® rapid labeling results in signal that is robust and specific. Confocal image of U2OS cells expressing HaloTag®-NLS (nuclear localization sequence) and labeled with the HaloTag® TMR Ligand using the rapid labeling protocol clearly shows a strong fluorescent signal that is restricted to the nucleus. Panels (left to right) show fluorescence, DIC image and overlay. The image was acquired on a confocal microscope equipped with fluorophore-appropriate filter sets and an environmental chamber in which the cells remained at 37°C + CO2 throughout imaging.
Live-Cell Labeling (No-Wash Protocol)
This protocol is intended for labeling live cells with Janelia Fluor® 549 HaloTag® Ligand or Janelia Fluor® 646 HaloTag® Ligand. These ligands have been demonstrated for use in super-resolution microscopy and single-molecule tracking approaches.
Materials To Be Supplied By The User
- Chambered cover glass with cells expressing HaloTag® fusion protein
- Complete culture medium appropriate for your cells, at 37°C
- Culture medium, lacking phenol red at 37°C (optional)
- DMSO
- Confocal microscope or wide-field fluorescent microscope equipped with appropriate filter sets and/or lasers
- 37°C + CO2 cell culture incubator
No-Wash Labeling Procedure
- Resuspend Janelia Fluor® 549 HaloTag® Ligand in 37.8µl of DMSO and Janelia Fluor® 646 HaloTag® Ligand in 35.5µl of DMSO. This will create a 200µM stock solution that is recommended for single-use only. Storage in DMSO is not recommended and will result in reduced stability of these ligands.
- Prepare a 1:200 (1µM final) dilution of each HaloTag® Ligand in warm culture medium just prior to addition to cells. This is a 5X working stock solution.
- Label cells by replacing one-fifth of the existing volume of medium with the 5X HaloTag® Ligand working stock solution, and mix gently. This results in the recommended final labeling concentration of 200nM.
- Incubate for 15 minutes or longer in a 37°C + CO2 cell culture incubator.
- Replace the medium with an equal volume of fresh warm culture medium. Optional: Replace the medium with an equal volume of fresh warm culture medium lacking phenol red. Use of medium lacking phenol red may improve imaging.
- Transfer to a microscope and capture images.
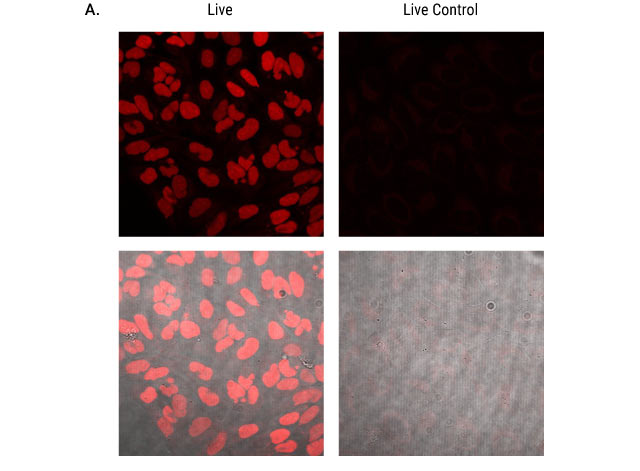
Figure 6: Live cell labeling of U2OS cells expressing nuclear HaloTag® protein. Parental U2OS cells and U2OS cells expressing HaloTag® containing a nuclear localization sequence were adhered to glass-bottom chamber slides and labeled with 200nM Janelia Fluor® 549 HaloTag® Ligand or Janelia Fluor® 646 HaloTag® Ligand for 15 minutes. Cells were imaged with 561nm laser excitation for Janelia Fluor® 549 HaloTag® Ligand (Panel A) and 637nm laser excitation for Janelia Fluor® 646 HaloTag® Ligand (Panel B). In HaloTag-expressing cells, labeling is restricted to the nucleus. Parental cells (live control in figure) show no labeling. The top row of each panel is fluorescent signal only. The bottom row shows transmitted images collected at the same respective laser excitation with the fluorescence images overlaid. Images were collected using a Nikon Eclipse Ti confocal microscope equipped with NIS Elements software and a 40X plan fluor oil immersion objective.
Fluorescence-Activated Cell Sorting (FACS®)
Materials Required
- large-well format plate of HaloTag®-expressing cells and negative control cells that do not express the HaloTag® fusion protein
- complete culture medium appropriate for your cells at 37°C
- for adherent cells: 1X PBS (pH 7.5) and 1X PBS containing 3mM EDTA or trypsin solution at 37 °C (see Step 2 below)
- cell sorting machine with appropriate filter sets and lasers for ligand used
- 37°C + CO2 cell culture incubator
Fluorescence-Activated Cell Sorting Procedure
- Label cells expressing HaloTag® protein with ligand of choice. Recommended controls: (a) Label cells that are not expressing HaloTag® protein to assess background with chosen ligand; (b) prepare unlabeled cells expressing HaloTag® protein to assess background from endogenous cell fluorescence.
- For adherent cells with HaloTag® fusion protein expressed on the cell surface: Rinse cells twice with an equal volume of 1X PBS, incubate in 1X PBS with 3mM EDTA at 37°C for 5 minutes, and then gently scrape the cells off of the plate.
For adherent cells with HaloTag® fusion protein expressed inside the cell: Suspend cells using trypsin-containing solution in a manner appropriate for the cell line. - Collect, pellet, count and resuspend cells in medium at 37°C to a concentration of 0.5 to 1 × 106 cells/ml.
- Sort cells.
- Place sorted cells in 37°C + CO2 incubator using a format appropriate for later application.
- Transfer to a microscope and image, and perform SDS-PAGE, or relabel cells (e.g., with a different ligand color) and image.
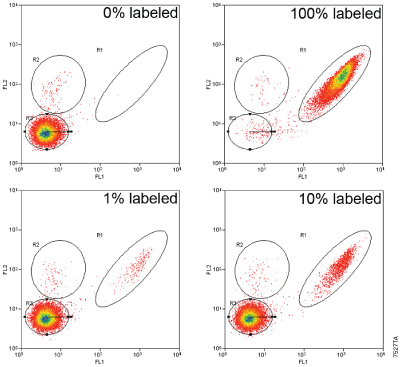
Figure 7. Successful FACS® of labeled HaloTag®-expressing cells. U2OS cells stably expressing HaloTag® 2 protein fused to three copies of a nuclear localization sequence were labeled with the HaloTag® R110Direct™ Ligand as described in Technical Manual #TM260, and known numbers (1%, 10% or 100% labeled) were sorted from nonlabeled cells. FACS® was performed using a MoFlo® instrument with approximately 20,000 cells represented on each graph. R1 indicates cells labeled with R110, R2 are dead cells (by propidium iodide), and R3 indicates unlabeled cells. Results show that labeled cells are successfully sorted from nonlabeled cells.
Fixed-Cell Labeling Protocol
This protocol is intended to serve as a guide to fix cells expressing a HaloTag® fusion protein. The covalent bond that forms between the ligand and HaloTag® protein during live-cell labeling allows you to subsequently fix, permeabilize and wash the cells under stringent conditions without significant loss of the specific fluorescent signal. We recommend the use of paraformaldehyde (PFA) as a fixative because it crosslinks proteins in cells and at the membrane and has the added benefit of reducing cell loss from the growth surface. Fixed cells can be treated with detergents, such as Triton® X-100, to further eliminate nonspecific labeling and permeabilize cells for downstream immunocytochemical applications. The conditions here are sufficient to permeabilize the plasma membrane. Alternative or additional detergents might be necessary to permeabilize other structures.
Materials Required
- HaloTag® pHT2 Vector or HaloTag® Flexi® Vectors, desired ligands, and protocol #TM260
- 4% paraformaldehyde/0.2M sucrose/1X PBS (pH 7.5)
- 1X PBS buffer (pH 7.5)
- 1X PBS + 0.1% Triton® X-100
- confocal microscope or wide-field fluorescent microscope equipped with appropriate filter sets and lasers
Fixed-Cell Labeling Procedure
- Follow Steps 1–5 of Rapid Labeling or No-Wash Labeling protocol to label cells, if desired.
- Replace the medium with an equal volume of warm 4% paraformaldehyde/0.2M sucrose/1X PBS (pH 7.5), and incubate for 10 minutes at room temperature.
- Replace fixative with an equal volume of 1X PBS + 0.1% Triton®X-100, and incubate for 10 minutes at room temperature.
- Replace the detergent-containing solution with an equal volume of 1X PBS.
- Transfer to a microscope and capture images, or proceed to the next section for an immunocytochemistry protocol.
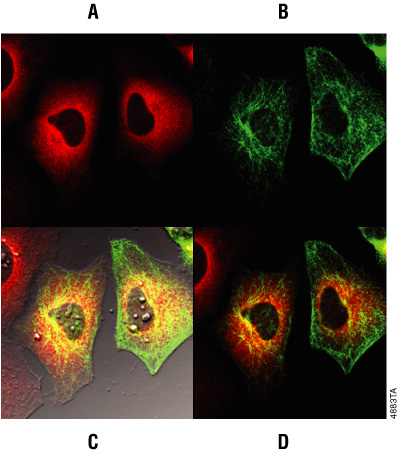
Figure 8. Multiplexing HaloTag® labeling with antibody staining. HeLa cells transiently transfected with the p65-HaloTag® construct and labeled with TMR Ligand were fixed and stained with mouse Anti-βIII Tubulin Antibody followed by incubation with Alexa Fluor® 488-conjugated goat anti-mouse IgG (Molecular Probes). Images were generated on an Olympus FV500 confocal microscope in sequential mode using appropriate filter sets. Panel A. TMR fluorescence. Panel B. Alexa Fluor® 488 fluorescence. Panel C. Overlay of Alexa Fluor® 488 and TMR fluorescence and transmitted light. Panel D. Overlay of Alexa Fluor® 488 and TMR fluorescence.
Immunocytochemistry Using the Anti-HaloTag® Polyclonal Antibody
This protocol is intended to serve as a guide for immunocytochemistry using Anti-HaloTag® Polyclonal Antibody. This antibody is a purified rabbit polyclonal antibody raised against the HaloTag® protein. The antibody was purified using Protein G affinity resin and has been shown to detect HaloTag® fusion proteins in both immunocytochemistry and Western blot applications with high sensitivity and specificity. Further, this antibody labels HaloTag® fusion proteins independently of HaloTag® ligands. As these labels do not interfere with one another, it is possible to colabel HaloTag® fusion proteins with a fluorescent ligand and the anti-HaloTag® pAb in conjunction with an anti-rabbit secondary antibody bearing a spectrally distinct fluorescent tag (Figure 9).
Materials Required
- 1X PBS buffer (pH 7.5)
- fluorophore-conjugated anti-rabbit secondary antibody of choice
- PBS + 2% donkey serum + 0.01% Triton® X-100
- PBS + 1% donkey serum
- confocal or wide-field fluorescent microscope equipped with appropriate filter sets and lasers
- PBS + 0.1% sodium azide (optional, for storage)
Immunocytochemistry Using the Anti-HaloTag® Polyclonal Antibody Procedure
- Cells expressing a HaloTag® fusion protein, whether labeled or unlabeled, should be fixed and permeabilized as described in Steps 2–5 above.
- Replace the 1X PBS with an equal volume of PBS + 2% donkey serum + 0.01% Triton® X-100, and block for 1 hour at room temperature.
- Dilute the Anti-HaloTag® pAb 1:500 in PBS + 1% donkey serum, to final labeling concentration of 2μg/ml.
- Replace the blocking solution with the antibody solution, and incubate for 1 hour at room temperature.
- Wash cells twice with PBS + 1% donkey serum for 10 minutes at room temperature.
- Dilute the secondary antibody according to the manufacturer's recommendations in PBS + 1% donkey serum.
- Replace the wash solution with the secondary antibody solution, and incubate for 30 minutes at room temperature.
- Wash cells twice with PBS + 1% donkey serum for 10 minutes each wash at room temperature.
- Replace wash solution with 1X PBS.
- Transfer to a microscope, and capture images. Store cells in PBS + 0.1% sodium azide.
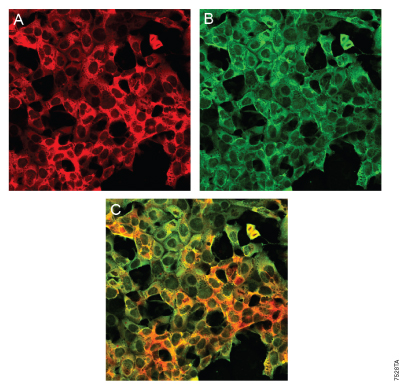
Figure 9. Distinct labeling of a single HaloTag® 7 fusion by a HaloTag® Ligand and Anti-HaloTag® pAb. HEK 293 cells stably transfected with a p65-HaloTag® 7 construct (p65 is a cytoplasmic signaling protein) were labeled with HaloTag® TMR Ligand and then processed for immunocytochemistry using the Anti-HaloTag® pAb. The secondary antibody used was Alexa Fluor® 488-conjugated anti-rabbit-IgG (Jackson ImmunoResearch). Distinct labeling is demonstrated by the fact that the red fluorescence associated with specific HaloTag® TMR Ligand labeling (Panel A) does not prevent green fluorescence associated with specific Anti-HaloTag® pAb labeling (Panel B). Merging the fluorescent images (Panel C) confirms that the HaloTag® fusion protein is labeled specifically (restricted to the cytoplasm), as expected. Images were collected on an Olympus FV500 confocal microscope in sequential mode using appropriate filter sets.
Multiplexing HaloTag® Protein, Other Reporters and/or Antibodies in Imaging Experiments
HaloTag® Technology can simplify multicolor/multiplex labeling experiments. The HaloTag® protein is not an intrinsically fluorescent protein (IFP), and the choice of fluorescent labels can be made after creating the HaloTag® fusion protein. This feature allows flexibility in experimental design for multicolor labeling as well as immunocytochemistry experiments.
To demonstrate this flexibility, we labeled cells expressing HaloTag®-α-tubulin fusion protein with the TMR Ligand or the diAcFAM Ligand and processed the cells for ICC using Anti-βIII Tubulin mAb (Cat.# G7121) and Alexa Fluor® 488 or Cy®3-conjugated secondary antibodies (Figure 10). All HeLa cells expressed βIII-tubulin in the cytoplasm. The HaloTag®-α-tubulin reporter was localized to the cytoplasm in the subpopulation of successfully transfected cells.
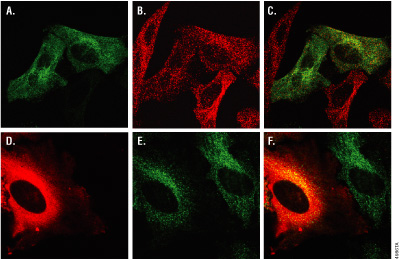
Figure 10. Multiplexing HaloTag® labeling and immunocytochemistry. HeLa cells were transfected with HaloTag®-α-tubulin fusion protein, labeled with HaloTag® diAcFAM (Panels A–C) or TMR Ligand (Panels D–F), washed and fixed as described in Los et al. 2005. Cells were immunolabeled with Anti-βIII Tubulin mAb (1:5,000 dilution, Cat.# G7121) followed by labeling with an Alexa Fluor® 488-conjugated secondary antibody (Panels A–C) or Cy®3-conjugated secondary antibody (Panels D–F). Panels A and D show cells the HaloTag® Ligands only; Panel A, diAcFAM Ligand; Panel D, TMR Ligand. Panels B and E show labeling for βIII tubulin. Panels C and F show the overlay of HaloTag® protein and βIII tubulin labeling.
Additional Resources for HaloTag® Interchangeable Labeling Technology
Technical Bulletins and Manuals
TM260 HaloTag® Technology: Focus on Imaging Technical Manual
TM254 Flexi® Vector Systems Technical Manual
Citations
Schröder, J. et al. (2009) In vivo labeling method using genetic construct for nanoscale resolution microscopy. Biophysical J. 96, L1–L3.
Traditionally light microscopy resolution has been limited by the diffraction of light. However several new technologies have emerged that partially overcome that limitation. One of these, stimulated emission depletion (STED) microscopy, is now commercially available and has been integrated into confocal microscope platforms. Because STED depends on fluorescent markers that fulfill specific spectroscopic needs, its uses have been limited. The authors of this paper demonstrate successful high resolution of β1-integrin-HaloTag®-fusion protein imaging using STED microscopy. This was possible because HaloTag® technology allows researchers to create a reporter that can be labeled with STED-optimized fluorescent tags.
PubMed Number: 19134467
Svendsen, S. et al. (2008) Spatial separation and bidirectional trafficking of proteins using a multi-functional reporter. BMC Cell Biology 9, 17.
This paper demonstrates use of HaloTag® technology to study expression and trafficking of an integrin-HaloTag® fusion protein. The authors fused the HaloTag® reporter protein to a truncated integrin. They then labeled live cells with different in a pulse-chase format using a green cell-impermeant ligand (Alexa Fluor® 488) followed by a cell-permeant TMR Ligand (red). This allowed direct monitoring of protein trafficking because pools of the same transmembrane protein were labeled with distinct fluorescent ligands.
PubMed Number: 18384686
Los, G.V. et al. (2008) HaloTag: A novel protein labeling technology for cell imaging and protein analysis. ACS Chemical Biology 3, 373–82.
The authors of this study describe the HaloTag® reporter system and use it to characterize a protein known to influence DNA transcription. The researchers create one genetic construct and demonstrate its use in determining protein localization and dynamics through imaging as well as biologically relevant interactions with a protein partner and DNA promoter.
PubMed Number: 18533659
Monster Green® Fluorescent Protein phMGFP Vector
With the discovery of intrinsically fluorescent proteins (IFPs) such as the green fluorescent protein (GFP) and the subsequent creation of a full-color spectrum of these IFPs, researchers can now fuse a protein of interest to IFPs with a variety of properties. IFPs are commonly used reporter molecules that can be visualized without cell lysis using standard fluorescence microscopy. They are often used to monitor gene expression but also can be used to monitor intracellular protein trafficking by creating C- and N-terminal protein fusions.
The Monster Green® Fluorescent Protein (Cat.# E6421) is an improved synthetic version of the green fluorescent protein gene originally cloned from Montastrea cavernosa (Great Star Coral). The synthetic gene (hMGFP) expresses a 26kDa protein that shows improved fluorescence intensity compared to the native gene. Peak excitation occurs at 505nm, and peak emission occurs at 515nm. Standard FITC filters may be used to visualize hMGFP fluorescence. The hMGFP gene is codon optimized and cleared of most consensus sequence transcription factor binding sites to ensure reliability and high levels of expression.
The Monster Green® Fluorescent Protein encoded by the hMGFP gene is an ideal fluorescent reporter, providing high-level fluorescence and reduced cytotoxicity. Monster Green® Protein generally fluoresces at least 20% brighter than other commercially available green fluorescent proteins (GFPs) and also reduces cytotoxicity, offering flexibility when working with transient and stable expression assays (Figure 11).
Additional Resources for the Monster Green® Fluorescent Protein
Technical Bulletins and Manuals
TB320 Monster Green® Fluorescent Protein phMGFP Vector Technical Bulletin
Citations
Inman, M. et al. (2004) Identification of a novel bovine herpesvirus 1 transcript containing a small open reading frame that is expressed in trigeminal ganglia of latently infected cattle. J. Virol. 78, 5438–47.
A fusion protein construct was made using the Monster Green® Fluorescent Protein phMGFP Vector and the PCR-amplified Open Reading Frame E (ORF-E) from Bovine herpesvirus 1. Transfected human neuroblastoma (SK-N-SH) cells, rabbit skin cells and bovine kidney cells were visualized using an Olympus FV500/BX60 confocal microscope with 488nm excitation laser and 522nm emission filter set. The ORF-E-MGFP protein was localized in discrete domains within the nucleus of Neuro-2A and SK-N-SH cells. In rabbit skin cells and bovine kidney cells the ORF-E-MGFP protein was detected in the cytoplasm and nucleus.
PubMed Number: 15113922
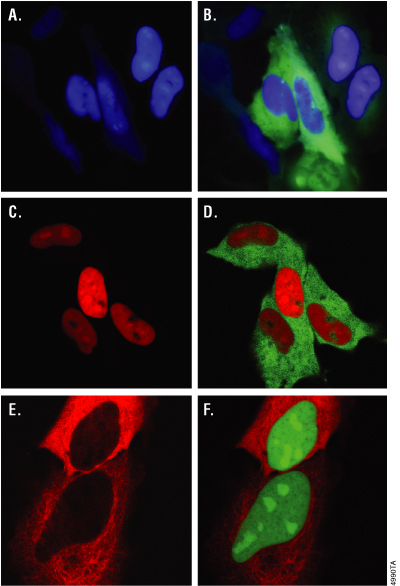
Figure 11. Expression of Monster Green® Fluorescent Protein in HeLa Cells. HeLa cells were transiently co-transfected with either HaloTag®-(NLS)3 plus hMGFP-α-tubulin (Panels A–D) or HaloTag®-α-tubulin plus hMGFP-(NLS)3 (Panels E and F). Twenty-four hours later, cells expressing HaloTag®-(NLS)3 were incubated with either 25μM Coumarin Ligand (Cat.# G8581, Panels A and B) or 5μM HaloTag® TMR Ligand (Panels C and D) for 15 minutes; cells expressing HaloTag®-α-tubulin were incubated with 5μM TMR Ligand for 15 minutes at 37°/5%CO2 (Panels E and F). Cells were washed and incubated for 30 minutes. In Panels A and B, cells were imaged with an Olympus IX81 epifluorescent microscope equipped with Chroma filter sets (#31000 DAPI for the Coumarin Ligand and #41001 FITC for hMGFP), a Hamamatsu Orca CCD camera and environmental controls. Images in Panels C–F were captured with an Olympus FV500 confocal attachment with sequential two-laser scanning and filters appropriate for TMR and FITC fluorescence.
Antibodies and Other Cellular Markers
Promega offers a variety of antibodies for detecting markers of apoptosis, assessing activation of cellular signaling pathways and monitoring indicators of cell type. These antibodies include antibodies raised against phosphorylated proteins, including kinases as well as antibodies against growth factors and growth-factor receptors.
Antibodies and Markers of Apoptosis
In situ Marker for Caspase-3: FITC-VAD-FMK
CaspACE™ FITC-VAD-FMK In Situ Marker (Cat.# G7461) is a fluorescent analog of the pan caspase inhibitor Z-VAD-FMK (carbobenzoxy-valyl-alanyl-aspartyl-[O-methyl]-fluoromethylketone). The fluorescein isothiocyanate (FITC) group has been substituted for the carbobenzoxy (Z) N-terminal blocking group to create the fluorescent apoptosis marker. This structure allows delivery of the inhibitor into the cell where it irreversibly binds to activated caspases. The FITC label allows for a single-reagent addition to assay for caspase activity in situ. The FITC-VAD-FMK Marker is supplied as a 5mM solution in DMSO and is intended for in situ monitoring of caspase activity by fluorescence detection. The suggested concentration for use in anti-Fas-treated Jurkat cell culture is 10µM.
Method for Detecting Apoptosis in Jurkat Cells
Materials Required
- CaspACE™ FITC-VAD-FMK In Situ Marker (Cat.# G7461, G7462)
- poly-l-lysine-coated slides
- anti-Fas mAb (Clone CH-11 MBL International Cat.# SY-100)
- PBS
- formalin
- mounting medium
- fluorescence microscope
Protocol
- Seed Jurkat cells at 1 × 105 cells/ml and grow in RPMI-1640 + 10% FBS in a 37°C, 5% CO2 incubator for 2–3 days until they reach a density of 5 × 105 cells/ml.
- Prepare poly-l-lysine-coated slides. Coat each chamber of multichamber slides with 0.01% poly-l-lysine solution. When partially dry, rinse the slides in NANOpure® water and then air-dry. Poly-l-lysine-coated slides can be prepared in advance and stored at 4°C for up to 7 days before use.
- To induce apoptosis, add anti-Fas mAb (Clone CH-11, MBL International Cat.# SY-100) to a final concentration of 0.1µg/ml. Do not add to controls. Incubate for 3–4 hours at 37°C.
- Add CaspACE™ FITC-VAD-FMK In Situ Marker to the Jurkat cells at a final concentration of 10µM. Protect cells from light and incubate for 20 minutes in the incubator. Keep cells protected from light for the remaining steps.
- Centrifuge at 300 × g for 5 minutes.
- Wash cells with PBS, then centrifuge at 300 × g for 5 minutes.
- Suspend cells in PBS to 1.5 × 106 cells/ml.
- Add cells to poly-l-lysine-coated slides and incubate at room temperature for 5 minutes to allow the cells to adhere to the poly-l-lysine.
- Fix in 10% buffered formalin for 30 minutes at room temperature (protected from light).
- Rinse 3 times for 5 minutes each time in PBS.
- Add mounting medium and coverslips to the slides. Analyze under a fluorescence microscope.
Additional Resources for the CaspACE™ FITC-VAD-FMK In situ Marker
Technical Bulletins and Manuals
9PIG746 CaspACE™ FITC-VAD-FMK In Situ Marker Product Information
Citations
Rouet-Benzineb, P. et al. (2004) Orexins acting at native OX1 receptor in colon cancer and neuroblastoma cells or at recombinant OX1 receptor suppress cell growth by inducing apoptosis. J. Biol. Chem. 279, 45875–86.
Human colon cancer (HT29-D4) cells were analyzed for activated caspases using the CaspACE™ FITC-VAD-FMK In Situ Marker. HT29-D4 cells (7 × 105) were cultured in the presence or absence of 1µM orexins, peptide growth inhibitors. Cells were washed, and bound CaspACE™ FITC-VAD-FMK In Situ Marker was visualized by confocal microscopy.
PubMed Number: 15310763
Detecting Active Caspase-3 Using an Antibody
Anti-ACTIVE® Caspase-3 pAb is intended for use as a marker of apoptosis; it specifically stains apoptotic human cells without staining nonapoptotic cells. All known caspases are synthesized as pro-enzymes activated by proteolytic cleavage. Anti-ACTIVE® Caspase-3 pAb is an affinity-purified rabbit polyclonal antibody directed against a peptide from the p18 fragment of human caspase-3. The antibody is affinity purified using a peptide corresponding to the cleaved region of caspase-3.
General Immunochemical Staining Protocol
Materials Required
- Anti-ACTIVE® Caspase-3 pAb
- prepared, fixed samples on slides
- Triton® X-100
- PBS
- blocking buffer (PBS/0.1% Tween® 20 + 5% horse serum)
- donkey anti-rabbit Cy®3 conjugate secondary antibody (Jackson Laboratories Cat.# 711-165-152)
- mounting medium
- humidified chamber
Protocol
- Permeabilize the fixed cells by incubating in PBS/0.2% Triton® X-100 for 5 minutes at room temperature. Wash three times in PBS, in Coplin jars, for 5 minutes at room temperature.
- Drain the slide and add 200µl of blocking buffer (PBS/0.1% Tween® 20 + 5% horse serum). Use of serum from the host species of the conjugate antibody (or closely related species) is suggested. Lay the slides flat in a humidified chamber and incubate for 2 hours at room temperature. Rinse once in PBS.
- Add 100µl of the Anti-ACTIVE® Caspase-3 pAb diluted 1:250 in blocking buffer. Prepare a slide with no Anti-ACTIVE® Caspase-3 pAb as a negative control. Incubate slides in a humidified chamber overnight at 4°C.
- The following day, wash the slides twice for 10 minutes in PBS, twice for 10 minutes in PBS/0.1% Tween® 20 and twice for 10 minutes in PBS at room temperature.
- Drain slides and add 100µl of donkey anti-rabbit Cy®3 conjugate diluted 1:500 in PBS. (We recommend Jackson ImmunoResearch Cat.# 711-165-152.) Lay the slides flat in a humidified chamber, protected from light, and incubate for 2 hours at room temperature. Wash twice in PBS for 5 minutes, once in PBS/0.1% Tween® 20 for 5 minutes and once in PBS for 5 minutes, protected from light.
- Drain the liquid, mount the slides in a permanent or aqueous mounting medium and observe with a fluorescence microscope.
Additional Resources for the Anti-ACTIVE® Caspase-3 pAb
Technical Bulletins and Manuals
9PIG748 Anti-ACTIVE® Caspase-3 pAb Product Information
Citations
Debacq-Chainiaux, F. et al. (2005) Repeated exposure of human skin fibroblasts to UVB at subcytotoxic level triggers premature senescence through the TGF-β1 signaling pathway. J. Cell Science 118, 743–58.
The authors of this study developed a model for UVB-induced premature senescence of skin human diploid fibroblasts. Markers of senescence were expressed in the system, and the authors were able to detect a common mitochondrial DNA 4,977bp deletion that is associated with oxidative damage. After a series of ten UVB stresses, total RNA was prepared from the cells using the RNAgents® Total RNA Isolation System and used for RT-PCR to detect differentially expressed genes. Forty-four stress or senescence-associated genes were identified that were differentially expressed between UVB irradiated and untreated cells including c-fos, c-jun, insulin-like growth factor binding protein 3, several HSPs, genes involved in protection from oxidative stress, and the type II receptor of TGF-β. The Anti-ACTIVE® Caspase-3 pAb was used to assess whether the UVB treatment or incubation with TGF-β1 led to apoptosis in this system.
PubMed Number: 15671065
Using an Antibody Against a Cleaved Caspase-3 Substrate (Anti-PARP p85 Fragment pAb)
Poly (ADP-ribose) polymerase (PARP), a nuclear enzyme involved in DNA repair, is a well-known substrate for caspase-3 cleavage during apoptosis. Anti-PARP p85 Fragment pAb is a rabbit polyclonal antibody specific for the p85 fragment of PARP that results from caspase cleavage of the 116kDa intact molecule and thus provides an in situ marker for apoptosis. Each batch of antibody is tested for use in immunostaining applications and contains sufficient antibody for 50 immunocytochemical reactions at a working dilution of 1:100.
General Immunocytochemistry Protocol
Materials Required
- Anti-PARP p85 Fragment pAb
- cells fixed on slides
- PBS
- blocking buffer (PBS/0.1% Tween® 20 + 5% horse serum)
- donkey anti-rabbit biotin conjugate (Jackson Cat.# 711-065-152) or donkey anti-rabbit Cy®3 conjugate (Jackson Cat.# 711-165-152)
- H2O2 (if using biotin conjugate)
- DAB solution (if using biotin conjugate)
- ultrapure water
- humidified chamber
- peroxidase-labeled streptavidin (eg., KPL Cat.# 14-300-00, diluted 1µg/ml in PBS)
Protocol
- Permeabilize cells fixed on slides in 0.2% Triton® X-100/PBS for 5 minutes at room temperature.
- Wash in 1X PBS in coplin jars for 5 minutes at room temperature. Repeat twice for a total of 3 washes.
- Drain the slides and add blocking buffer (PBS/0.1% Tween® 20 + 5% normal serum). Cover cells with blocking buffer (200µl per slide). Lay the slides flat in a humidified chamber and incubate for 2 hours at room temperature.
- Rinse once in PBS.
- Add 100µl of the Anti-PARP p85 Fragment pAb diluted in blocking buffer. We recommend a starting dilution of 1:100. Include a slide with no Anti-PARP p85 Fragment pAb as a negative control. Incubate slides in a humidified chamber overnight at 4°C.
- The following day, wash the slides twice for 10 minutes in 1X PBS, twice for 10 minutes in PBS/0.1% Tween® 20, and twice for 10 minutes in 1X PBS at room temperature.
- If the secondary antibody is a horseradish peroxidase (HRP) conjugate, block endogenous peroxidases by incubating with 0.3% hydrogen peroxide for 4–5 minutes at room temperature. If you are using a different method of detection with a secondary antibody, proceed to Step 9.
- Wash in 1X PBS in Coplin jars for 5 minutes. Repeat twice for a total of 3 washes.
- Drain slides and add 100–200µl of diluted secondary antibody to each slide. We recommend donkey anti-rabbit biotin conjugate (Jackson Cat.# 711-065-152) or donkey anti-rabbit Cy®3 conjugate (Jackson Cat.# 711-165-152) diluted 1:500 in PBS/0.1% Tween® 20. Lay the slides flat in a humidified chamber and incubate for 2 hours at room temperature.
- Wash several times in 1X PBS.
- For the biotin conjugate, drain the slides and add 100–200µl of Streptavidin-HRP solution to each slide. Lay the slides flat in a humidified chamber and incubate for 45 minutes at room temperature. For HRP-conjugated secondary antibodies, proceed to Step 13. For other secondary antibodies, proceed to Step 15.
- Wash in 1X PBS in Coplin jars for 5 minutes. Repeat twice for a total of 3 washes.
- Add 100–200µl of freshly made diaminobenzidine (DAB) solution to each slide. Lay the slides flat and incubate for ~10 minutes at room temperature.
- Rinse the slides in NANOpure® water. Bleach is frequently used to inactivate the DAB before disposal; however, local requirements for hazardous waste should be followed.
- Drain the liquid and mount the slides in a permanent or aqueous mounting medium (slides mounted in 70% glycerol can be stored for several weeks at 4°C or –20°C).
Method for Staining Postnatal Day 0 Mouse Brain, Paraffin-Embedded Sections (all steps performed at room temperature)
Materials Required
- Anti-PARP p85 Fragment pAb
- paraffin-embedded, fixed sample
- Hemo-De® (Fisher Scientific) or xylene
- ethanol (100, 95 and 70%)
- PBS
- Triton® X-100
- H2O2
- biotin-conjugated donkey anti-rabbit pAb
- RTU ABC reagent (Vector Laboratories)
- DAB substrate kit (Vector Laboratories)
- VECTASHIELD® DAPI anti-fade Reagent (Vector Laboratories)
Protocol
- Embed tissue in paraffin after fixation in 4% paraformaldehyde. Six micron sections are used for this protocol.
Note: Best results will be obtained if the animal is perfused with fix and postfixed after dissection. - Deparaffinize by washing tissue 3 times for 5 minutes each in Hemo-De® (Fisher Scientific) or xylene. Rinse tissue sections for 2 minutes in 100% ethanol. Transfer sections to 95% ethanol for 2 minutes, then transfer them to 70% ethanol for 2 minutes. Finally, rinse tissue sections 2 times for 2 minutes each in PBS.
- Permeabilize for 10 minutes in PBS + 0.1% Triton® X-100.
- Wash sections 2 times for 5 minutes each in PBS.
- Block endogenous peroxide activity by incubating sections in 0.3% H2O2 in PBS for 30 minutes.
- Wash sections 2 times for 5 minutes each in PBS.
- Block for 45 minutes in PBS + 5% donkey serum.
- Incubate with Anti-PARP p85 Fragment pAb diluted 1:50 in PBS + 1.0 % donkey serum for 60 minutes.
- Wash sections 3 times for 5 minutes each in PBS.
- Incubate with biotin-conjugated donkey anti-rabbit pAb (Jackson ImmunoResearch) diluted 1:500 in PBS for 60 minutes.
- Wash sections 3 times for 5 minutes each in PBS.
- Incubate in R.T.U. (Ready-To-Use) ABC reagent (Vector Laboratories) for 60 minutes.
- Wash sections 3 times for 5 minutes each in PBS.
- Develop with DAB substrate kit (Vector Laboratories) for 10 minutes.
- Wash 3 times for 5 minutes each in water.
- Mount in VECTASHIELD® + DAPI anti-fade reagent (Vector Laboratories).
- Analyze samples immediately using a fluorescence microscope.
Additional Resources for the Anti-PARP p85 Fragment pAb
Technical Bulletins and Manuals
TB273 Anti-PARP p85 Fragment pAb Technical Bulletin
Citations
Davidson, B. et al. (2003) Expression and activation of the nerve growth factor receptor TrkA in serous ovarian carcinoma. Clin. Cancer Res. 9, 2248–59.
Anti-PARP p85 Fragment pAb was used to stain human peritoneal and pleural effusions.
PubMed Number: 12796393
O'Brien, M.A., Moravec, R.A. and Riss, T. (2001) Poly (ADP-ribose) polymerase cleavage monitored in situ in apoptotic cells. Biotechniques 30, 886–91.
The authors demonstrate specificity of an affinity-purified polyclonal antibody to the p85 fragment of PARP with Western blots that show that the antibody recognizes the 85kDa (p85) fragment of PARP but not full-length PARP.
PubMed Number: 11314271
Antibodies and Cellular Markers for Studying Cell Signaling Pathways
Promega provides a variety of phosphorylation-specific antibodies for studying cell signaling pathways. Brief descriptions of these products are provided below.
Anti-ACTIVE® Phosphorylation-Specific Antibodies
The Anti-ACTIVE® phosphorylation-specific antibodies were developed to provide an accurate measure of enzyme activation. These antibodies specifically recognize the active, phosphorylated form of a given kinase. The Anti-ACTIVE® Antibodies are raised against phosphorylated peptide sequences present in the activating loop of a number of protein kinases. Whether used in Western analysis, immunocytochemistry or immunohistochemical staining, the Anti-ACTIVE® MAPK, JNK, p38 and CaM KII Antibodies will recognize only the active form of the enzyme.
Anti-ACTIVE® MAPK pAb, Rabbit (pTEpY)
This antibody is an affinity-purified polyclonal antibody that specifically recognizes the dually phosphorylated, active form of MAPK. The antibody is raised against a dually phosphorylated peptide sequence representing the catalytic core of the active ERK enzyme and recognizes the active forms of ERK1, ERK2 and ERK7.
Anti-ACTIVE® JNK pAb, Rabbit (pTPpY)
Anti-ACTIVE® JNK pAb is an affinity-purified polyclonal antibody that recognizes the dually phosphorylated, active form of cJun N-terminal protein Kinase (JNK). Anti-ACTIVE® JNK pAb is raised against a dually phosphorylated peptide sequence representing the catalytic core of the active JNK enzyme. The antibody recognizes the active forms of JNK1, JNK2, and JNK3 isoforms.
Anti-ACTIVE® p38 pAb, Rabbit, (pTGpY)
Anti-ACTIVE® p38 Ab, Rabbit, is an affinity-purified polyclonal antibody that recognizes the active form of p38 kinase. The Anti-ACTIVE® p38 pAb is raised against the dually phosphorylated peptide sequence representing the catalytic core of the active p38 enzyme. The Anti-ACTIVE® p38 pAb recognizes the active forms of p38α, γ, and δ isoforms.
Additional Resources for the Anti-ACTIVE® Antibodies
Technical Bulletins and Manuals
TB262 Anti-ACTIVE® MAPK, p38 and JNK Polyclonal Antibodies and Anti-ACTIVE® Qualified Secondary Antibody Conjugates
Citations
Hsu, C.Y. et al. (2004) Characterization of active mitogen-activated protein kinase in ovarian serous carcinomas. Clin. Can. Res. 10, 6432–6.
The Anti-ACTIVE® MAPK polyclonal antibody was used to immunohistochemically stain and type patient ovarian serous carcinomas using paraffin-fixed tissue sections on tissue microarrays. Western blots were also performed on tissue lysates using a 1:3,000 dilution of the antibody.
PubMed Number: 15475429
Le'Negrate, G. et al. (2003) Downregulation of caspases and Fas ligand expression, and increased lifespan of neutrophils after transmigration across intestinal epithelium. Cell Death Differ. 10, 153–62.
Anti-ACTIVE® JNK pAb was used in immunoblot analysis of human polymorphonuclear leukocyte protein lysates.
PubMed Number: 12700643
Aballay, A. et al. (2003) Caenorhabditis elegans innate immune response triggered by Salmonella enterica requires intact LPS and is mediated by a MAPK signaling pathway. Curr. Biol. 13, 47–52.
Activation of the p38 homolog in the worm was monitored by Western analysis using the Anti-ACTIVE® p38 pAb.
PubMed Number: 12526744
Marker Antibodies
Anti-βIII Tubulin mAb
Anti-βIII Tubulin mAb (Cat.# G7121) is a protein G-purified IgG1 monoclonal antibody (from clone 5G8) raised in mice against a peptide (EAQGPK) corresponding to the C-terminus of βIII tubulin. It is directed against βIII tubulin, a specific marker for neurons. The major use of this antibody is for labeling neurons in tissue sections and cell culture. The antibody performs in frozen and paraffin-embedded sections of rat brain, cerebellum and spinal cord, human and rat fetal CNS progenitor cell cultures and adult human paraffin-embedded brain (Figure 12).
- Immunogen: Peptide corresponding to the C-terminus (EAQGPK) of βIII tubulin.
- Antibody Form: Mouse monoclonal IgG1 (clone 5G8), 1mg/ml in PBS containing no preservatives.
- Specificity: Cross-reacts with most mammalian species. Does not label nonneuronal cells (e.g., astrocytes).
- Suggested Working Dilutions: Immunocytochemistry (1:2,000), Immunohistochemistry (1:2,000), Western blotting (1:1,000 dilution).
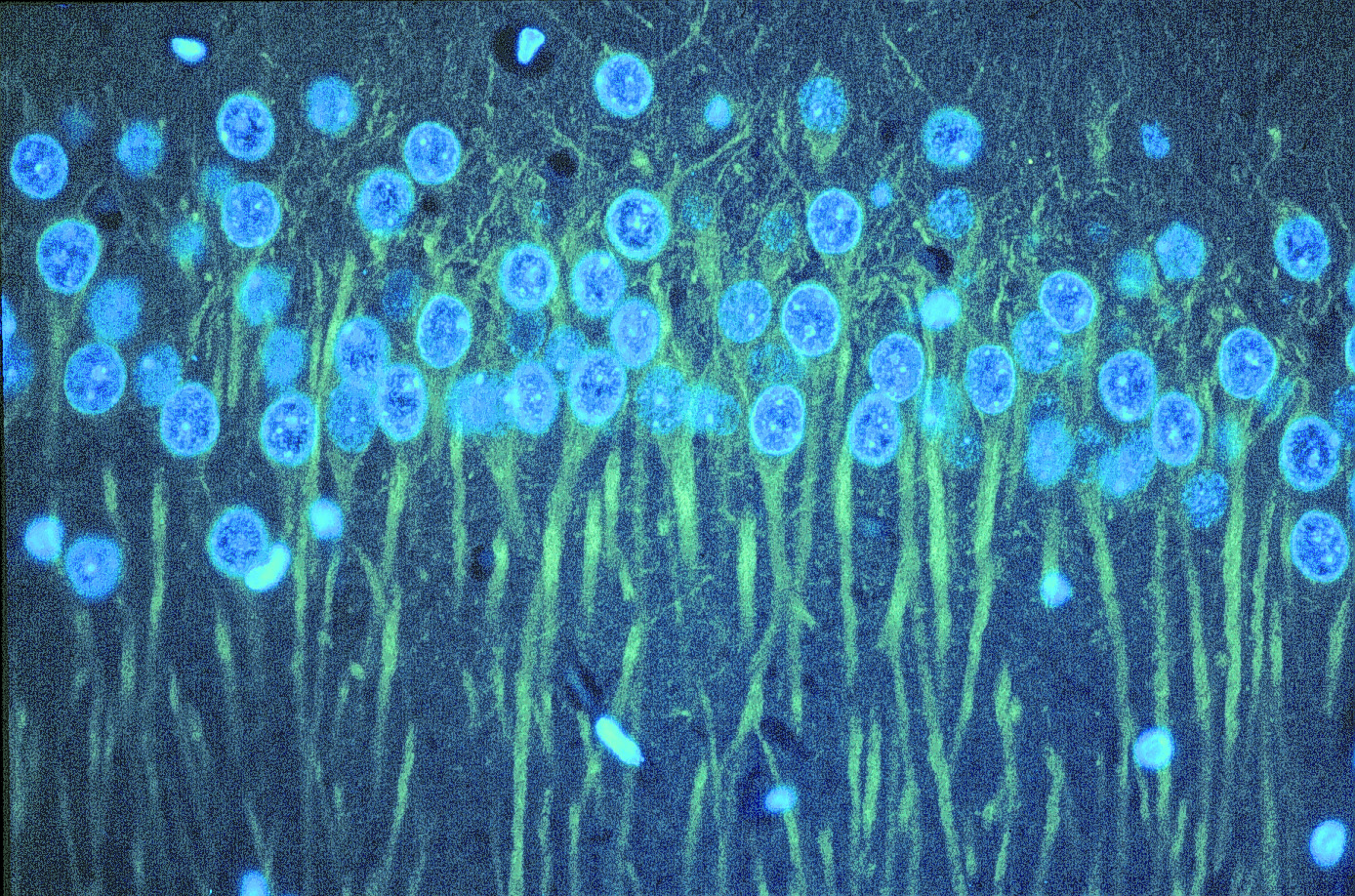
Figure 12. Immunostaining for βIII tubulin in rat cerebellum using Anti-βIII Tubulin mAb. Paraffin-embedded rat brain section double-immunofluorescence-labeled with the primary antibody and detected using an anti-mouse Cy®3-conjugated secondary antibody (yellow-green). Nuclei were stained with DAPI (blue). Protocols developed and performed at Promega.
Additional Resources for Anti-βIII Tubulin mAb
Promega Publications
CN012 Perform multicolor live- and fixed-cell imaging applications with the HaloTag® Interchangeable Labeling Technology
Citations
Brunelli, G. et al. (2005) Glutamatergic reinnervation through peripheral nerve graft dictates assembly of glutamatergic synapses at rat skeletal muscle. Proc. Natl. Acad. Sci. USA 102, 8152–7.
PubMed Number: 15937120
Walker, K. et al. (2001) mGlu5 receptors and nociceptive function II. mGlu5 receptors functionally expressed on peripheral sensory neurons mediate inflammatory hyperalgesia. Neuropharmacology 40, 10–9.
Rat skin sections were subjected to immunohistochemistry with the Anti-βIII Tubulin mAb to detect metabolic glutamate receptor expressing neurons. Twenty micron sections were fixed in acetone, permeabilized with 0.1% Triton® X-100 and incubated with the Anti-βIII Tubulin mAb at a final concentration of 1μg/ml.
PubMed Number: 11077066
Related Products
HaloTag® Technology
HaloTag is a powerful technology with applications for protein purification, protein localization, trafficking and turnover as well as protein interactions and super-resolution microscopy.
References
- Check, E. (2005) Ethicists urge caution over emotive power of brain scans. Nature 435, 254–5.
- Eisenstein, M. (2006) Helping cells to tell a colorful tale. Nature Methods 3 647–55.
- Lang, C. et al. (2006) HaloTag®: A new versatile reporter gene system in plant cells. J. Exp. Bot 57 2985–92.
- Los, G.V. et al. (2005) HaloTag™ Interchangeable Labeling Technology for cell imaging and protein capture. Cell Notes 11, 2–6.
- McKenna, N.M. and Wang, Y-l. (1989) Culturing cells on the microscope stage. Methods in Cell Biol. 29, 1–12. San Diego, CA: Academic Press.
- Owen, R.M. et al. (2002) Synthesis and applications of end-labeled neoglycopolymers. Org. Lett. 4 2293–6.
- Shinohara, H. and Matsubayahsi, Y. (2007) Functional immobilization of plant receptor-like kinase onto microbeads toward receptor array construction and receptor-based ligand fishing. Plant J. 52 175–84.
- Stephens, D.J. and Allan, V.J. (2003) Light microscopy techniques for live cell imaging. Science 300, 82–86.
- Sullivan, K.F. and Kay, S.A. (1999) Preface. Methods in Cell Biol. 58, xv–xvi.
- Wang, Y-l. (1989) Fluorescent analog cytochemistry: Tracing functional protein components in living cells. Methods in Cell Biol. 29, 1–12.
- Zhang, Y. et al. (2006) HaloTag® protein-mediated site-specific conjugation of bioluminescent proteins to quantum dots. Angew. Chem. Int. Ed. Engl. 45 4936–40.