Protease Digestion for Mass Spectrometry
This guide contains protocols for protein digestion with trypsin, trypsin/lys-C or alternative proteases.
For ordering information on the products discussed here, please visit our Proteases product pages.
Mass Spec Analysis
Mass spectrometry is a leading analytical method in proteomics (Mann et al., 2001). Mass spectrometry is used for protein identification, the study of protein:protein interactions, characterization of post-translational modifications (e.g., phosphorylation, glycosylation, methylation and acetylation) and protein quantification (relative and absolute).
These applications primarily employ bottom-up proteomics, where proteins of interest are digested with an enzyme such as trypsin and the resulting peptides are analyzed by mass spectrometry. Bottom-up proteomics focuses on the analysis of protein mixtures after enzymatic digestion of the proteins into peptides. The resulting complex mixture of peptides is analyzed by reverse-phase liquid chromatography (RP-LC) coupled to tandem mass spectrometry (MS/MS). Identification of peptides and subsequently proteins is completed by matching peptide fragment ion spectra to theoretical spectra generated from protein databases.
Trypsin has become the gold standard for protein digestion to peptides for shotgun proteomics. Trypsin is a serine protease. It cleaves proteins into peptides with an average size of 700-1500 daltons, which is in the ideal range for MS (Laskay et al., 2013). It is highly specific, cutting at the carboxyl side of arginine and lysine residues. The C-terminal arginine and lysine peptides are charged, making them detectable by MS. Trypsin is highly active and tolerant of many additives.
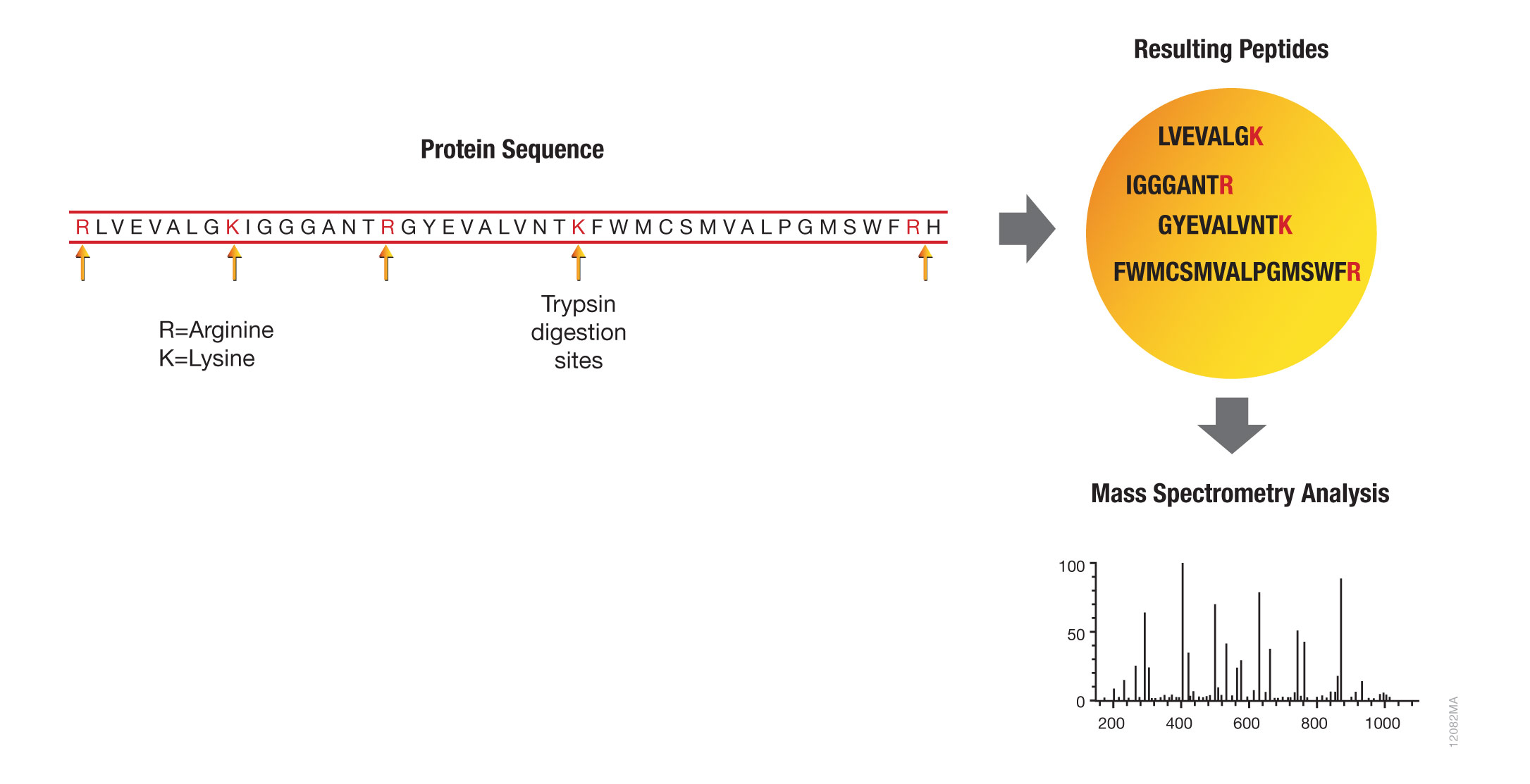
Figure 1. Bottom-up proteomic analysis with trypsin.
Trypsin Digestion Protocols
Stringent specificity of trypsin activity is crucial for protein identification. However, specificity can be compromised by autolysis, which generates pseudotrypsin with a broadened specificity and chymotrypsin-like activity (Keil-Dlouha et al., 1971). Autolysis can result in additional peptide fragments that could interfere with database analysis and protein identification.
Autolysis is suppressed by reductive methylation of lysine residues, yielding a highly stable molecule (Rice et al., 1977). For maximum specificity, Promega offers Sequencing Grade Modified Trypsin (Cat. # V5111 and V5117), which undergoes reductive methylation and TPCK treatment to improve trypsin cleavage specificity. TPCK inactivates chymotrypsin activity. To further improve proteolytic efficiency, Promega developed a more active Trypsin Gold, Mass Spectrometry Grade (Cat. # V5280). More information and a detailed protocol are available in the Trypsin Gold, Mass Spectrometry Grade Technical Bulletin #TB309.
In-Gel Protein Digestion
Numerous protocols for in-gel protein digestion exist (Flannery et al., 1989; Shevchenko et al., 1996; Rosenfeld et al., 1992). The following procedure has been used successfully by Promega scientists. For a more streamlined protocol, see the protocol for In-Gel Digestion of Proteins Using Trypsin and ProteaseMAX™ Surfactant, Trypsin Enhancer in the ProteaseMAX™ Surfactant, Trypsin Enhancer, Technical Bulletin #TB373.
Materials Required:
- Trypsin Gold, Mass Spectrometry Grade (Cat.# V5280) and protocol
- SimplyBlue™ SafeStain (Invitrogen Cat.# LC6060)
- trifluoroacetic acid (TFA)
- acetonitrile (ACN)
- 200mM NH4HCO3 buffer (pH 7.8)
- 50mM acetic acid
- NANOpure® water
- ZipTip® scx pipette tips (Millipore Cat.# ZTSCXS096) or ZipTip® C18 pipette tips (Millipore Cat.# ZTC18S096)
- 0.5ml microcentrifuge tubes prewashed twice with 50% ACN/0.1% TFA
- Speed Vac® concentrator
- Separate protein samples by electrophoresis on an SDS-Tris-Glycine gel.
Note: Other gel systems and staining reagents can be used for in-gel digestions but should be tested to ensure compatibility with the protein of interest and detection system being used. - Rinse the gel for 5 minutes with NANOpure® water. Repeat this wash twice for a total of three washes. Stain for 1 hour in SimplyBlue™ SafeStain at room temperature with gentle agitation. When staining is complete, discard the staining solution.
- Destain the gel for 1 hour in NANOpure® water at room temperature with gentle agitation. When destaining is complete, discard the solution.
- Using a clean razor blade, cut the protein bands of interest from the gel, eliminating as much polyacrylamide as possible. Place the gel slices in a 0.5ml microcentrifuge tube prewashed twice with 50% ACN/0.1% TFA.
- Destain the gel slices with 0.2ml of 100mM NH4HCO3/50% ACN for 45 minutes at 37°C. Repeat this destaining step once.
- Dehydrate the gel slices for 5 minutes at room temperature in 100μl of 100% ACN. After dehydration, the gel slices will be much smaller than their original size and will be whitish or opaque in appearance.
- Dry the gel slices in a Speed Vac® concentrator for 10–15 minutes at room temperature.
- Resuspend the Trypsin Gold at 1μg/μl in 50mM acetic acid, and then dilute in 40mM NH4HCO3/10% ACN to 20μg/ml. Preincubate the gel slices in a minimal volume (10–20μl) of the trypsin solution at room temperature (do not exceed 30°C) for 1 hour. The slices will rehydrate during this time. If the gel slices appear white or opaque after one hour, add an additional 10–20μl of trypsin and incubate for another hour at room temperature.
- Add enough digestion buffer (40mM NH4HCO3/10% ACN) to completely cover the gel slices. Cap the tubes tightly to avoid evaporation. Incubate overnight at 37°C.
- Incubate the gel slices with 150μl of NANOpure® water for 10 minutes, with frequent vortex mixing. Remove and save the liquid in a new microcentrifuge tube.
- Extract the gel slice twice with 50μl of 50% ACN/5% TFA (with mixing) for 60 minutes each time at room temperature.
- Pool all extracts (from Steps 10 and 11), and dry in a Speed Vac® concentrator at room temperature for 2–4 hours (do not exceed 30°C).
- Purify and concentrate the extracted peptides using ZipTip® pipette tips (Millipore Corporation) following the manufacturer’s directions.
- The peptides eluted from the ZipTip® tips are now ready for mass spectrometric analysis.
In-Solution Protein Digestion
Typically, proteins are reduced and then alkylated to allow immediate access of trypsin to internal cleavage sites. This warrants high protein sequence coverage during mass spectrometry analysis (Wilkinson, 1986). If high sequence coverage is not required, reduction and alkylation steps can be omitted. Note: To digest a nonreduced protein, begin this protocol at Step 3.
- Dissolve the target protein in 8M urea/50mM Tris-HCl (pH 8) [or 50mM ammonium bicarbonate (pH 7.8)]/ 5mM DTT, and incubate at 37°C for 1 hour.
Note: Protein denaturation with guanidine chloride (GuCl) is not recommended because even low GuCl concentrations inhibit trypsin. - Add iodoacetamide to a final concentration of 15mM, and incubate for 30 minutes in the dark at room temperature.
- Dilute the reaction with three volumes of 50mM Tris-HCl (pH 8) [or 50mM ammonium bicarbonate (pH 7.8)] to reduce the urea concentration to 2M. For digestion of native proteins, dissolve the protein in 50mM NH4HCO3 (pH 7.8) or 50mM Tris-HCl (pH 8) without urea.
- Add Trypsin Gold, Mass Spectrometry Grade, to a final protease:protein ratio of 1:100 to 1:20 (w/w). Incubate overnight at 37°C.
- Terminate the digestion by adding TFA or formic acid to a final concentration of 1%, and analyze the sample by high performance chromatography (HPLC) or liquid chromatography-mass spectrometry (LC/MS).
Trypsin Gold, Mass Spec Grade Protocol
More information and a detailed protocol for use of Trypsin Gold, Mass Spec Grade are available in Technical Bulletin #TB309.
Affinity Tag In Vitro Pull-Down Assay with Trypsin Digestion and Protein Analysis
Markillie and colleagues described a simple exogenous protein complex purification and identification method that can be easily automated (Markillie et al., 2005). The method uses MagneHis™ Ni Particles (Cat.# V8560, V8565) to pull down target proteins, followed by denaturing elution, trypsin digestion and mass spectrometry analysis (Figure 2).
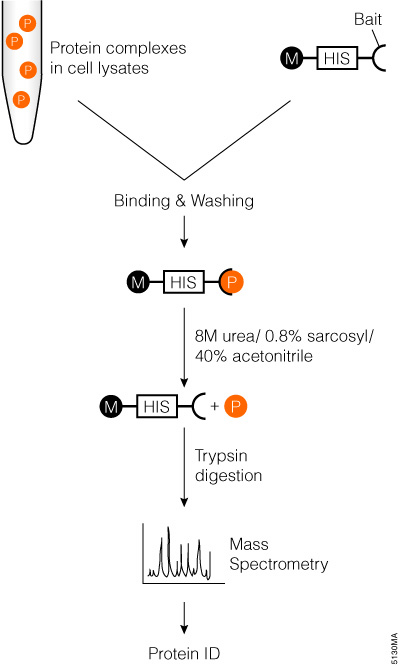
Figure 2. Schematic diagram of affinity tag in an vitro pull-down assay with trypsin digestion and mass spectrometry analysis.
Trypsin/Lys-C Mix, Mass Spec Grade
Trypsin/Lys-C, Mass Spec Grade (Cat.# V5071), is a trypsin preparation with the highest proteolytic efficiency. It is a mixture of Trypsin Gold, Mass Spectrometry Grade, and rLys-C, Mass Spec Grade. Proteolysis with Trypsin/Lys-C Mix, Mass Spec Grade, generates tryptic peptides (i.e., peptides with C-terminal arginine and lysine residues). With the conventional trypsin digestion protocol (i.e., overnight incubation under nondenaturing conditions), Trypsin/Lys-C Mix improves protein digestion by eliminating the majority of missed cleavages (Figure 3).
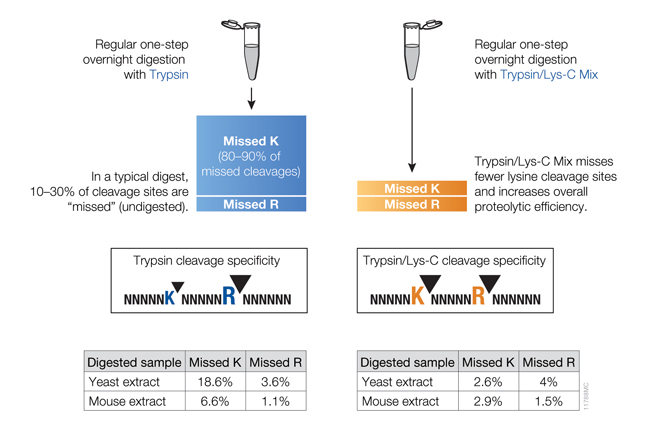
Figure 3. Side-by-side comparison of cleavage sites missed by trypsin or the Trypsin/Lys-C Mix using a conventional digestion protocol.
Tightly folded proteins represent a particular challenge; these proteins are resistant to proteolysis due to inaccessibility of internal cleavage sites for trypsin. Using Trypsin/Lys-C Mix helps overcome this challenge by enabling a two-step digestion protocol (Figure 4), which utilizes the tolerance of Lys-C protease to protein denaturing conditions. In step 1, a protein is denatured with 8M urea. Under these conditions, Lys-C remains active and digests a protein into relatively large fragments. In the second step, the digestion mixture is diluted fourfold to reduce urea concentration to 2M. This reactivates trypsin and allows complete proteolysis.
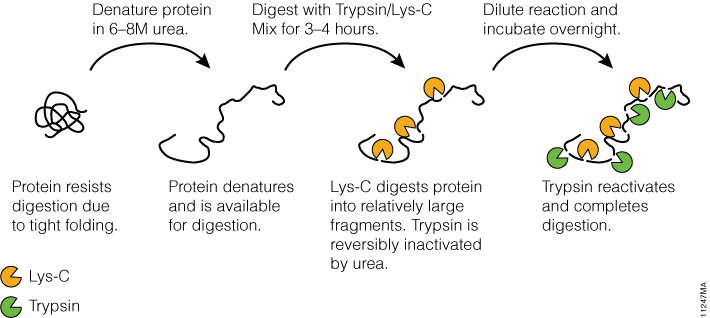
Figure 4. Digesting difficult-to-digest proteins using a specialized two-step procedure with Trypsin/Lys-C Mix.
Trypsin/Lys C Protocol
More information and protocol details are available in the Trypsin/Lys-C Mix, Mass Spec Grade, Technical Manual #TM390.
Alternative Proteases
The use of trypsin in bottom-up proteomics may impose certain limits in the ability to grasp the full proteome. Tightly-folded proteins can resist trypsin digestion. Post-translational modifications (PTMs) present a different challenge for trypsin because glycans often limit trypsin access to cleavage sites, and acetylation makes lysine and arginine residues resistant to trypsin digestion.
To overcome these problems, the proteomics community has begun to explore alternative proteases to complement trypsin. However, protocols, as well as expected results generated when using these alternative proteases have not been systematically documented.
Giansanti et. al. (2016) optimized protocols for six alternative proteases that have already shown promise in their applicability in proteomics, namely chymotrypsin, Lys-C, Lys-N, Asp-N, Glu-C and Arg-C have been created.
There are certain instances when trypsin does not provide adequate proteolysis. For example, many membrane proteins have limited number of tryptic cleavage sites. In other cases, distribution of tryptic cleavage sites is suboptimal, resulting in peptides that are too long or too short for mass spectrometry analysis. Promega offers various alternative proteases that complement trypsin and allow efficient protein analysis with mass spectrometry. Figure 5 highlights the benefits of the alternative protease chymotrypsin for protein mass spectrometry analysis.
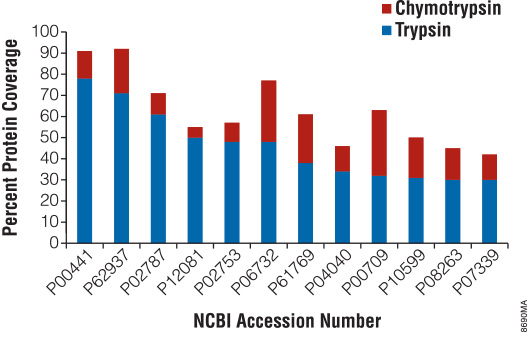
Figure 5. Increased protein coverage using both trypsin and chymotrypsin.
Site-Specific Proteases
Arg-C, Sequencing Grade (Cat.# V1881), also known as clostripain, is an endopeptidase that cleaves at the C-terminus of arginine residues, including sites next to proline. Arg-C activity is optimal at pH 7.6–7.9.
Asp-N, Sequencing Grade (Cat.# V1621), is an endoproteinase that hydrolyzes peptide bonds at the N-terminal side of aspartic acid residues. Asp-N activity is optimal at pH 4.0–9.0.
rAsp-N, Mass Spec Grade (Cat.# VA1160), is a recombinant protease that was cloned from Stenotrophomonas maltophilia and purified from E. coli.
Chymotrypsin, Sequencing Grade (Cat.# V1061), is a serine endoproteinase derived from bovine pancreas. The protease preferentially hydrolyzes at the carboxyl side of aromatic amino acids: tyrosine, phenylalanine and tryptophan. Chymotrypsin activity is optimal at pH 7.0–9.0.
Glu-C, Sequencing Grade (Cat.# V1651), is a serine protease that cleaves specifically at the C-terminal side of glutamic acid residues in the presence of ammonium bicarbonate or ammonium acetate. In phosphate buffers, cleavage also occurs at aspartic acid residues. Glu-C activity is optimal at pH 4.0–9.0.
rLys-C, Mass Spec Grade (Cat.# V1671), is recombinant endoproteinase Lys-C expressed in E. coli. The sequence origin of rLys-C is Protease IV from Pseudomonas aeruginosa. Similarly to native Lys-C, rLys-C cleaves at the C-terminus of lysine residues with exceptional specificity and retains proteolytic activity under denaturing conditions. rLys-C activity is optimal at pH 8.0–9.0.
Endoproteinase Lys-C, Mass Spec Grade (Cat.# VA1170), is a highly-purified serine protease that hydrolyzes specifically at the carboxyl side of lysines. Lys-C retains proteolytic activity under strong protein denaturing conditions.
Nonspecific Proteases
Elastase (Cat.# V1891) is a serine protease that preferentially cleaves at the C-terminus of alanine, valine, serine, glycine, leucine or isoleucine residues. Elastase activity is optimal at pH 9.0.
Pepsin (Cat.# V1959) preferentially cleaves at the C-terminus of phenylalanine, leucine, tyrosine and tryptophan residues. Pepsin activity is optimal at pH 1.0–3.0.
Thermolysin is a thermostable metalloproteinase. Thermolysin preferentially cleaves at the N-terminus of the hydrophobic residues leucine, phenylalanine, valine, isoleucine, alanine and methionine. The optimal digestion temperature range is 65–85°C. Thermolysin activity is optimal at pH 5.0–8.5.
| rLys-C | Lys-C | rLys-N | rAsp-N | Asp-N | Arg-C | Glu-C | |
| Cat.# | V1671 | VA1170 | VA1180 | VA1160 | V1621 | V1881 | V1651 |
| Source and Size | Pseudomonas aeruginosa, expressed in E. coli (27.7kDa) | Lysobacter enzymogenes (30kDa) |
Grifola frondosa (18kDa) | Stenotrophomonas maltophilia (25kDa) |
Pseudomonas fragi (24.5kDa) |
Clostridium histolyticum (Subunits: 45kDa and 12kDa) | Staphylococcus aureus V8 (27kDa) |
| Cleavage Sites | C-terminal of Lys. Does not cleave if Lys is followed by Pro. Asp or Glu at C-terminal side of Lys inhibits cleavage. | C-terminal of Lys. Does not cleave if Lys is followed by Pro. Asp or Glu at C-terminal side of Lys inhibits cleavage. | Cleaves at the N-terminus of Lysine. | Primarily on the N-terminal side of aspartic acid residues. Cleavage on the N-terminal side of glutamic acid residues can occur at a slower rate. | N-terminal of Asp. | C-terminal of Arg. Also cleaves at Lys albeit at lower efficiency. | C-terminal of Glu. Low level cleavages might occur at Asp residues too albeit at 100–300 fold lower efficiency. |
| Protease:Protein Ratio (w/w) | 1:20 to 1:50 | 1:20 to 1:100 | 1:20 to 1:100 | 1:10 to 1:100 | 1:20 to 1:200 | 1:20 to 1:350 | 1:20 to 1:200 |
| Optimal pH Range | pH 8–9 | pH 7–9 | pH 7–9 | rAsp-N has maximal activity at pH 8, but pH buffers ranging from 6–9 can be used. | pH 4–9 | pH 7.6–7.9 | pH 4–9 |
| Reaction Conditions | 50–100mM Tris-HCl (pH 8) or 50mM NH4HCO3 (pH 7.8). Digestion at 37°C for 2–18 hours. | Incubate at 37°C for 2–18 hours. | Incubate at 37°C for 2–18 hours. | Incubate at 37°C for 60 minutes. | 50mM Tris-HCl (pH 8). Digestion 2–18 hours at 37°C. | 50mM Tris-HCl (pH 7.6-7.9), 5mM CaCl2, 2mM EDTA, >2mM DTT. Digestion 2–18 hours at 37°C. | 100mM NH4HCO3 (pH 7.8), 50-100 mM HCL (pH 8). Digestion 2–18 hours at 37°C. |
| Buffer Compatibility | Tris-HCl, NH4HCO3 | 50mM Tris (pH 8) | 50mM Tris (pH 8) | Ammonium acetate (pH 5-6), HEPES (pH 7), Tris (pH 8-9), Ammonium Bicarbonate (pH 8) | Tris-HCl, NH4HCO3 | Tris-HCl, NH4HCO3 | NH4HCO3, Ammonium acetate |
| In-Gel Digestion Compatibility | Yes | Not tested | Not tested | Not tested | Yes | Yes | Yes |
| ProteaseMAX™ Compatibility | Yes | Not tested | Not tested | Not tested | Yes | Yes | Yes |
| Notes | Inexpensive alternative to a native Lys-C protease. Similarly to a native protease, rLys-C tolerates high denaturing conditions such as 8M urea. Used to digest tightly folded proteolytically resistant proteins. Also used as a trypsin alternative if larger peptides are preferable for the analysis. If urea is used in protein sample preparation, avoid high temperature. High temperature induces protein carbamylation in the presence of urea. |
Tolerates high denaturing conditions such as 8M urea. Used to digest tightly folded proteolytically resistant proteins. Also used as a trypsin alternative if larger peptides are preferable for analysis. If urea is used in protein sample preparation, avoid high temperature. High temperature induces protein carbamylation in the presence of urea. | Lys-N is active in up to 6M urea, although activity will vary depending on concentration. If urea is used in protein sample preparation, avoid high temperature. High temperature induces protein carbamylation in the presence of urea. | rAsp-N, Mass Spec Grade, is lyophilized in Tris (pH 8.0) with NaCl and stabilizing sugars. Reconstitute in 50µl of ultrapure water and mix gently. Reconstituted rAsp-N can be stored at 4°C for at least 8 weeks. For longer storage, single-use aliquots can be stored at –65°C or below. Avoid freeze-thaw cycles. rAsp-N has a histidine tag that can be used to remove it from solution. | Can be used as a trypsin alternative to achieve better distribution of cleavage sites.100% activity retained in the presence of urea (up to 3.5 M), guanidine HCL (1M), SDS (up to 0,028%), ProteaseMax™ Surfactant (up to 0,026%), acetonitrile (up to 60%), EDTA (up to 2mM); DTT or ß-mercaptoethanol. | Used in analysis of histone modifications. Requires DTT, cysteine or other reducing agent and CaCl2 for activity. | Can be used as a trypsin alternative to achieve better distribution of cleavage sites. Glu-C activity and cleavage specificity is affected by buffer conditions. In ammonium bicarbonate and other nonphosphate buffers, Glu-C cleaves at C-term of Glu. Glu-C cleaves at C-term of Glu and Asp in phosphate buffer. |
More About Alternative Proteases
Article: Advantages to Using Arg-C, Elastase, Thermolysin and Pepsin for Protein Analysis
Alternative Proteases Webinar
You can learn more about use of alternative proteases to improve coverage in the On-demand Webinar: Bottoms Up: Improving Proteomic Sample Preparation with Recombinant Asp-N.
ProteaseMAX™ Trypsin Enhancer
ProteaseMAX™ Surfactant, Trypsin Enhancer, enables fast and efficient in-solution and in-gel protein digestion with proteases such as trypsin and Lys-C. The surfactant is an efficient protein-solubilizing agent. (Figure 6). As an additive with urea, ProteaseMAX™ Surfactant improves the solubilizing effects of urea.

Figure 6. Improved in-solution protein digestion due to protein denaturation using the ProteaseMAX™ Surfactant.
The ProteaseMAX™ Surfactant offers several advantages for in-gel protein digestion. First, this surfactant improves protein identification through enhanced protein digestion and peptide extraction (Figure 7). Second, the surfactant minimizes peptide adsorption to plasticware, which is the major cause of peptide loss during in-gel protein digestion. Third, ProteaseMAX™ Surfactant eliminates the need for post-digestion extraction and accelerates digestion. In the presence of ProteaseMAX™ Surfactant in-gel protein digestion is complete in a single 1-hour step.
ProteaseMAX™ Surfactant is an anionic surfactant that degrades over the course of a digestion reaction, generating degradation products that are innocuous to mass spectrometry. This feature eliminates the need for post-digestion degradation.

Figure 7. Example of improved protein identifications when using ProteaseMAX™ Surfactant, Trypsin Enhancer, for in-gel digestion of a complex protein sample.
In-Gel Digestion of Proteins Using Trypsin and ProteaseMAX™ Surfactant, Trypsin Enhancer
In-gel protein digestion saves time and labor. The digestion step is complete in 1 hour, and the ProteaseMAX™ Surfactant provides concurrent extraction of peptides from gels, eliminating the need for postdigestion peptide extraction. The surfactant also improves recovery of longer peptides that are typically retained in the gel using a standard extraction protocol. For a detailed protocol, refer to the ProteaseMAX™ Surfactant, Trypsin Enhancer, Technical Bulletin #TB373.
ProteaseMAX™ Protocol
For more information about the use of ProteaseMAX™ Surfactant and post-digestion peptide handling, refer to the ProteaseMAX™ Surfactant, Trypsin Enhancer, Technical Bulletin #TB373.
References
- Flannery, A.V. et al. (1989) "Proteolysis of proteins for sequencing analysis and peptide mapping." In: Proteolytic Enzymes: A Practical Approach. R.J. Beynon and J.S. Bond, eds., IRL Press, Oxford, 45.
- Giansanti, P. et al. (2016) Six alternative protease for mass spectrometry based proteomics beyond trypsin. Nat. Protocols 11, 993–6.
- Keil-Dlouha, V. et al. (1971) Proteolytic activity of pseudotrypsin. FEBS Lett. 16, 291–5.
- Laskay, U. et al. (2013) Proteome Digestion Specificity Analysis for the Rational Design of Extended Bottom-up and middle-down proteomics experiments. J. Proteome Res. 12, 5558–69.
- Mann, M. et al. (2001) Analysis of proteins and proteomes by mass spectrometry. Annu. Rev. Biochem. 70, 437–73.
- Rice, R.H. et al. (1977) Stabilization of bovine trypsin by reductive methylation. Biochim. Biophys. Acta 492, 316–21.
- Rosenfeld, J. et al. (1992) In-gel digestion of proteins for internal sequence analysis after one- or two-dimensional gel electrophoresis. Anal. Biochem. 203, 173–9.
- Shevchenko, A. et al. (1996) Mass spectrometric sequencing of proteins silver-stained polyacrylamide gels. Anal. Chem. 68, 850–8.