Scientific Issues with Analysis of Low Amounts of DNA
National Institute of Standards and Technology, Biochemical Science Division, Gaithersburg, Maryland, USA
Publication Date: 2010
Abstract
Faced with limited evidence that yield low amounts of DNA, forensic analysts will continually have to confront the question of how far to push DNA-testing techniques. Low copy number (LCN) analysis, also known as low template DNA (LT-DNA) testing, involves enhancing detection sensitivity usually through increasing the number of PCR cycles. Stochastic effects inherent with analysis of low amounts of DNA yield allele or locus drop-out. Additionally, increasing detection sensitivity can result in a greater potential for contamination or allele drop-in. Validation studies with replicate testing of low amounts of DNA were performed to assess the level of allele and locus drop-out and allele drop-in using 10, 30 and 100 picograms with several commercially available STR-typing kits under both standard and increased number of PCR cycles. The results with pristine, fully heterozygous samples demonstrate that a replicate testing approach can produce reliable information with single-source samples when consensus profiles are created.
Introduction
Due to the limited nature of biological evidence that may be recovered from some crime scene samples, decisions often have to be made whether or not to proceed with testing low amounts of DNA. With any scientific measurement, validation helps define limits of the techniques used. In the case of forensic DNA validation, sensitivity studies where one or more control DNA samples are diluted to low amounts enable a laboratory to establish at what point a detection technique cannot deliver reliable results. Repeated testing of replicated aliquots of the same diluted control DNA samples permits an evaluation of result reproducibility. This article will examine some simple validation experiments performed with low amounts of DNA and their implications.
Stochastic Effects during PCR Amplification
As forensic DNA analysts attempt to recover information from low amounts of DNA present in evidentiary samples, they will encounter stochastic or random sampling effects that occur in the early cycles of PCR amplification. When a limited number of DNA target molecules exist in a sample, the PCR primers used to amplify a specific region may not consistently find and hybridize to the entire set of DNA molecules present in the amplification reaction. With a heterozygous locus, where two alleles are present, unequal sampling of the alleles can result in failure to detect one or both of the alleles. Loss of a single allele is referred to as “allele drop-out” while loss of both alleles is termed “locus drop-out”.
Stochastic (random) variation is a fundamental physical law of the PCR amplification process when examining low amounts of DNA. Stochastic effects are manifest as a fluctuation of results between replicate analyses. In other words, amplifying the same DNA extract twice can result in different alleles being detected at a locus.
Since stochastic effects cannot be avoided when testing small quantities of DNA, there are essentially two schools of thought on how to handle these types of samples: 1) stop testing or interpreting data before you go low enough to be in the stochastic realm, or 2) try to limit the impact of the stochastic variation by additional testing and careful interpretation guidelines based on validation studies. Those who advocate the second approach usually enhance their method sensitivity, such as increasing the number of PCR cycles, to get as much out of the limited sample as possible. The “enhanced interrogation” approach typically involves replicate testing and the development of consensus profiles.
The “Stop Testing” Approach
The “stop testing” approach begs the question of how do you know that you are too low to obtain reliable results (i.e., information that accurately reflects the DNA sample). There are two primary points in the DNA-testing process where DNA reliability may be assessed: 1) at the DNA quantitation stage prior to performing PCR amplification of the short tandem repeat (STR) markers of interest, or 2) during examination of peak heights—and peak height ratios in heterozygous loci—in the STR profile obtained. An empirically determined threshold (usually termed a “stochastic threshold”) may be used at either the DNA quantitation or data interpretation stage to assess samples in the potential “danger zone” of unreliable results. For example, if the total amount of measured DNA is below 150pg, a laboratory may decide not to proceed with PCR amplification, assuming that allelic drop-out due to stochastic effects is a very real possibility. Alternatively, a laboratory may proceed with testing a low-level DNA sample, then evaluate the peak height signals and peak height ratios at heterozygous loci. Usually when peak height ratios for heterozygous loci in single-source samples dip below 60%, there is an indication that stochastic effects are significant, which would make it challenging to reliably pair alleles into major and minor genotypes with mixtures.
Since the advent of quantitative PCR (qPCR) assays, DNA quantitation tests have become more sensitive—enabling quantities as small as a few genomic copies to be detected. Use of qPCR assays, such as Quantifiler®(1) or Plexor® HY(2), can enable detection of minute amounts of DNA. However, it is important to keep in mind that qPCR also is subject to stochastic variation, especially on the low end of DNA quantity measurement. Thus, numbers in the low picogram range may not be reliable, and results with little or no “detectable” DNA may still amplify with STR kits(3).
In an early paper discussing stochastic effects and limitations of PCR assays, Walsh et al.(4) proposed avoiding stochastic effects by adjusting the number of PCR cycles in an assay so that the sensitivity limit is around 20 or more copies of target DNA. In other words, their goal was to enable a full DNA profile to be obtained reliably with approximately 125 pg of DNA. Below roughly that amount, allele and locus drop-out would be expected and partial DNA profiles would result. Depending on the STR-typing primer and DNA polymerase concentrations and fluorescent dye sensitivities, the number of PCR cycles typically is set by manufacturers in the range of 28–32 cycles.
The “Enhanced Interrogation” Approach
Improved sensitivity in a detection technique is usually a valuable asset to enable results to be obtained from limited biological evidence. Not satisfied with failure to obtain a result with low amounts of DNA template, some laboratories have decided to push the envelope and apply what could be termed “enhanced interrogation techniques”. In fact, DNA testing has been applied successfully down to the single cell level(5).
About a decade ago, the United Kingdom’s Forensic Science Service pioneered the application of low copy number (LCN) analysis through increasing the number of PCR cycles to improve DNA detection sensitivity(6)(7). Instead of the STR kit manufacturer’s recommended 28 cycles, which has a theoretical yield of 67 million copies for each target DNA sequence, an additional six cycles (34 total) were run to provide a theoretical yield of 4.3 billion copies or a 64-fold improvement in sensitivity. A more recent approach to high-sensitivity DNA testing uses only a three-cycle signal enhancement to provide a theoretical 16-fold improvement in sensitivity(8).
When pushing assay sensitivity through an increased number of PCR cycles, stochastic effects can become more evident. Figure 1 illustrates different types of stochastic effects that may be observed when performing PCR amplification from low amounts of DNA.
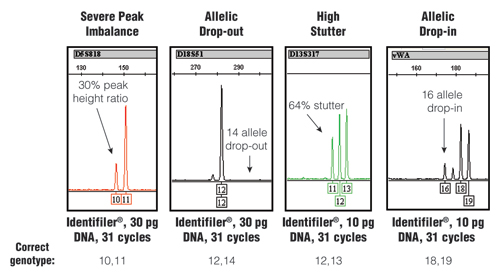 Figure 1. Stochastic effects that randomly occur when PCR amplifying low amounts of DNA using an increased number of PCR cycles.
Figure 1. Stochastic effects that randomly occur when PCR amplifying low amounts of DNA using an increased number of PCR cycles.
The STR-typing kit, amount of DNA and number of PCR cycles along with the correct genotype for each example are listed at the bottom. For further information, see www.cstl.nist.gov/biotech/strbase/LTDNA.htm
The stochastic variation observed with the test results shown in Figure 1 would produce an incorrect result if no further information was available. Loss of signal, such as in the D18S51 allelic drop-out example, would make a true “12,14” heterozygote appear as a “12,12” homozygote. Likewise, the gain of signal with the high stutter or allelic drop-in could make a true single-source sample appear to be a mixture. Thus, when using enhanced interrogation techniques, such as a higher number of PCR cycles, further testing measures are required to avoid reporting incorrect results.
Consensus Profiles Generated from Replicate Testing
To avoid or limit the possibility of getting the wrong answer when testing low amounts of DNA, replicate PCR amplifications are performed and a consensus profile developed(9). The standard practice is to PCR amplify either two or three aliquots of a DNA extract(6)(8). Alleles that occur more than once (i.e., repeat) in the obtained profiles are deemed “reliable” as they have been reproduced in separate DNA tests. Based on observations during validation studies, another layer of interpretation may be applied as well before the final consensus profile is reported. For example, specific loci, such as those larger in size, may exhibit a higher rate of allelic drop-out. When reporting results from these loci, a wildcard designation may be used in conjunction with a repeated single allele to account for potential allelic drop-out (e.g., “12,F” or “12,Z” instead of “12,12”).
While stochastic effects exist whenever low amounts of DNA are being examined with PCR, a replicate amplification approach with development of a consensus profile from the repeated alleles can produce reliable results (Figure 2). However, amplification results from a single test can be unreliable due to allelic drop-out or allelic drop-in as noted earlier. As seen in Figure 3, individual results from replicate tests may vary, but a combined consensus profile can generate an accurate answer when repeated alleles are recorded.
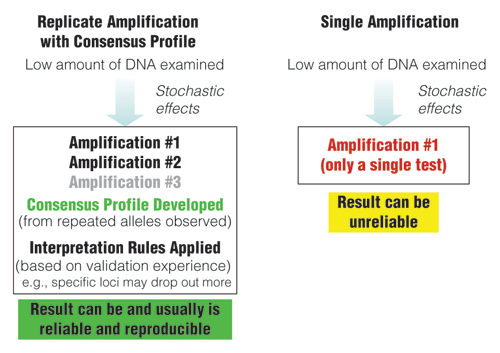 Figure 2. Comparison of approaches when examining low amounts of DNA.
Figure 2. Comparison of approaches when examining low amounts of DNA.
Replicate amplification with development of consensus profiles and application of interpretation rules based on validation experience can lead to reliable results.
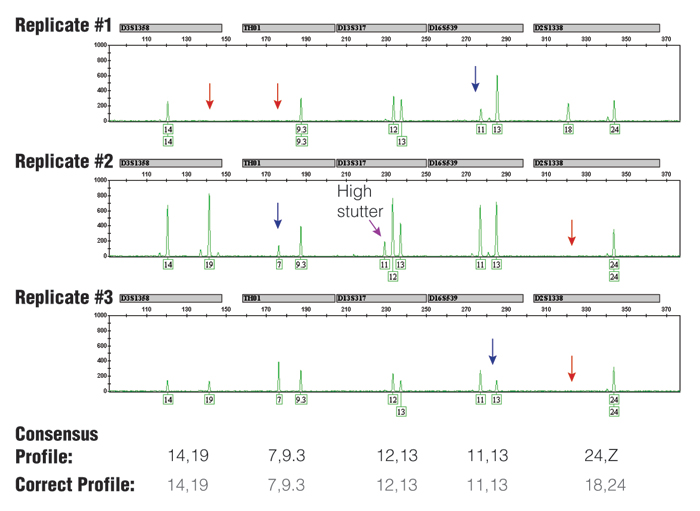 Figure 3. Three replicate PCR amplifications of a 10pg single-source DNA template using the Identifiler® kit (only green loci shown) and 31 cycles.
Figure 3. Three replicate PCR amplifications of a 10pg single-source DNA template using the Identifiler® kit (only green loci shown) and 31 cycles.
The consensus profile, produced by recording all alleles that occurred at least two out of three times, matched the correct profile (note that the consensus wildcard "Z" for D2S1338 appropriately covers the allele 18 that has dropped out twice). The red arrows indicate positions of allele drop-out, and the blue arrows indicate where severe peak height imbalance was observed.
NIST Data Collected using Low Amounts of DNA
To evaluate reliability of results with low levels of DNA template separately from the effects of DNA degradation or PCR inhibition that might be present in forensic specimens, validation experiments were conducted at the National Institute of Standards and Technology (NIST) with pristine DNA samples. Two single-source DNA samples, which were fully heterozygous at all of the tested loci, were quantified using the Quantifiler® kit (Applied Biosystems, Foster City, CA) using a calibration curve created with NIST SRM 2372 Component A (NIST, Gaithersburg, MD).
Following DNA quantification, dilutions were made to enable testing of 100pg, 30pg and 10pg of DNA template in each PCR. A total of 10 separate amplification reactions were performed for each sample to study the impact and value of replicate amplifications. While 10 replicates would not be practical to perform in a casework setting with limited forensic evidence, the extra studies are valuable in a validation context to examine if more than three replicates are helpful. Both the AmpFlSTR® Identifiler® kit (Applied Biosystems) and PowerPlex® 16 HS System (Promega Corporation, Madison, WI) were examined using half reactions to conserve costs and improve sensitivity. Identifiler® was tested at both 28 (standard) and 31 (enhanced) cycles. PowerPlex® 16 HS was tested at 31 (standard) and 34 (enhanced) cycles.
The resulting 240 electropherograms were examined to assess stochastic effects such as allelic drop-out and allelic drop-in. These electropherograms are available for review (as pdf files) at: www.cstl.nist.gov/biotech/strbase/LTDNA.htm. With 16 loci for each result, a total of 3,840 loci (7,680 alleles) potentially could be scored. In this analysis, all peaks above 50 relative fluorescence units (RFU) were called without an attempt to filter genotypes based on heterozygote peak height ratios.
Figure 4 illustrates the results for one of the tested samples with the PowerPlex® 16 HS System using 31 and 34 cycles. Note that use of a higher number of cycles (34 cycles) resulted in more correct genotypes as denoted by the green squares. The three extra PCR cycles improved the success rate for a correct heterozygous call from 69% (331/480 possible) to 91% (435/480 possible). With 34 cycles, allelic drop-out fell from 19% (92/480) to 8% (37/480), and locus drop-out fell from 12% (57/480) to <1% (4/480). Thus, boosting the cycle number did improve the sensitivity and overall success rate.
However, there were four instances of allele drop-in (Figure 4, black squares) when a higher number of PCR cycles were used, while none existed with the lower number of cycles. The potential occurrence of allele drop-in shows the importance of replicate amplifications and development of consensus profiles to avoid miscalls when utilizing enhanced interrogation techniques. Two of the allele drop-ins noted in Figure 4 are in FGA where the correct genotype is “21,25”, yet a “21,24,25” was called with the “24” likely coming from elevated stutter of the “25” allele. The other two drop-ins were at the N+4 stutter position in D5S818 and CSF1PO.
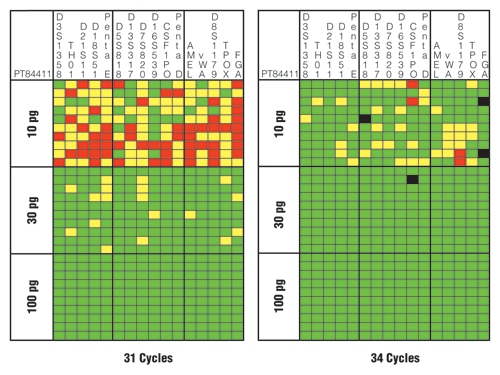 Figure 4. Sensitivity and performance summaries for PowerPlex® 16 HS System 31-cycle and 34-cycle data.
Figure 4. Sensitivity and performance summaries for PowerPlex® 16 HS System 31-cycle and 34-cycle data.
A horizontal slice represents information from a single sample, whereas a vertical slice represents the results from each of 10 replicates with three different DNA template amounts. Green squares indicate that the full correct type was observed for that locus and PCR replicate. Yellow squares denote allele drop-out where one of the expected alleles is missing. Red squares highlight locus drop-out where both expected alleles are missing. Black squares indicate allele drop-in where an incorrect allele is observed above a 50RFU analytical threshold.
It is probably worth noting that the other sample tested with the PowerPlex® 16 HS System only had one observed incidence of allele drop-in at a stutter position in D3S1358 (see www.cstl.nist.gov/biotech/strbase/LTDNA/PP34-summary.pdf). Likewise, the Identifiler® elevated cycle results had two incidences of allelic drop-in with one sample and four with the other sample but at different loci than seen with the PowerPlex® 16 HS System (see www.cstl.nist.gov/biotech/strbase/LTDNA/ID31-summary.pdf). Thus, each STR-typing kit will perform differently and needs to be internally validated for the specific conditions being examined in your laboratory. Certain loci are more prone to allele and locus drop-out, depending on the STR kit used.
In all cases, replicate testing and development of consensus profiles would have successfully excluded any incorrect calls due to allelic drop-in with the single-source samples examined in our study. Equally important is that across any group of three replicates, there was never an instance of an incorrect allele being called when two of the three replicates matched.
New STRBase Web Site on Low Template DNA
A new section of the NIST STRBase web site was launched in October 2009 following the International Symposium on Human Identification (ISHI) LCN Panel. This web site, which is available at: www.cstl.nist.gov/biotech/strbase/LTDNA.htm, contains three primary sections: 1) presentations, 2) NIST data from sensitivity studies, and 3) a listing of literature on the topic.
With the LTDNA web site launch, four presentations given at the ISHI 2009 meeting were included. The initial presentations by John Butler, Becky Hill, Theresa Caragine, and Charlotte Word hopefully will be added to in the future. These presentations are provided as pdf files, enabling easy access with Adobe Acrobat Reader.
Data from the experiments described earlier using Identifiler® and PowerPlex® 16 HS kits with 100pg, 30pg and 10pg and different numbers of PCR cycles are available to be viewed as pdf files. Two DNA samples that are heterozygous at all STR loci examined provide an opportunity to monitor peak imbalance and allelic drop-out under different conditions. Each sample, DNA amount and number of cycles was evaluated with 10 replicates. In total, there are 240 electropherograms to review along with summary graphs like that in Figure 4. These data help illustrate the stochastic variation observed when amplifying low amounts of DNA, including allele drop-out, allele drop-in, high stutter and heterozygote peak imbalance. We encourage laboratories to submit their validation data for inclusion on this community resource as suggested previously(10).
A listing of pertinent articles to help explain the issues involved with low template DNA testing are provided on the STRBase LTDNA web site and will continue to be updated over time. The articles are listed according to four categories in order to reflect their relative reliability in scientific terms: 1) peer-reviewed literature, 2) reports, 3) reviews, and 4) non-peer-reviewed literature. In science, as in other fields, not all information is equally authoritative or helpful. Thus, the literature on low template DNA analysis is broken into several categories on the STRBase web site to reflect the variation in scrutiny and support.
Concluding Remarks
Every lab faces samples with low amounts of DNA. Laboratories and DNA analysts need to choose whether or not to attempt an “enhanced interrogation technique” such as increasing the cycle number, desalting samples or higher CE injection. If such an approach is taken, validation studies need to be performed to develop appropriate interpretation guidelines and to assess the degree of variation that can be expected when analyzing low amounts of DNA.
Deciding where to stop testing or interpreting data can be challenging. Some laboratories stop testing based on a certain amount of input DNA, using validation data to underpin a quantitation threshold. Others set stochastic thresholds that are used during data interpretation to decide what STR-typing data are reliable (i.e., are not expected to have allelic drop-out at that locus). Performing experiments similar to those described in this article can help determine an appropriate stochastic threshold.
The next-generation STR kits, such as the PowerPlex® 16 HS, ESI 17 and ESX 17 Systems and AmpFlSTR® Identifiler® Plus and NGM kits, with their greater sensitivity and ability to overcome PCR inhibition, have the potential to make the current qPCR DNA quantitation kits obsolete as an appropriate gatekeeper to whether or not to continue with a low-level, compromised DNA sample. With greater power to get results comes greater responsibility to report reliable results. Careful validation studies and development of appropriate interpretation guidelines will continue to be essential as the forensic DNA community moves forward with caution and care in analysis of low amounts of DNA.
Acknowledgments
This work was funded in part by the National Institute of Justice (NIJ) through an interagency agreement 2008-DN-R-121 with the NIST Office of Law Enforcement Standards. Points of view in this document are those of the authors and do not necessarily represent the official position or policies of the U.S. Department of Justice. Certain commercial equipment, instruments and materials are identified in order to specify experimental procedures as completely as possible. In no case does such identification imply a recommendation or endorsement by the National Institute of Standards and Technology nor does it imply that any of the materials, instruments or equipment identified are necessarily the best available for the purpose.
Helpful Resources
Balding, D.J. and Buckleton, J. (2009) Interpreting low template DNA profiles. Forensic Sci. Int. Genet. 4, 1–10.
Budowle, B., Eisenberg, A.J. and van Daal, A. (2009) Validity of low copy number typing and applications to forensic science. Croat. Med. J. 50, 207–17.
Butler, J.M. (2010) Fundamentals of Forensic DNA Typing. Elsevier Academic Press, San Diego.
Gill, P., Puch-Solis, R. and Curran, J. (2009) The low-template-DNA (stochastic) threshold—its determination relative to risk analysis for national DNA databases. Forensic Sci. Int. Genet. 3, 104–11.
Article References
- Green, R.L. et al. (2005) Developmental validation of the Quantifiler™ real-time PCR kits for the quantification of human nuclear DNA samples. J. Forensic Sci. 50, 809–25.
- Krenke, B.E. et al. (2008) Developmental validation of a real-time PCR assay for the simultaneous quantification of total human and male DNA. Forensic Sci. Int. Genet. 3, 14–21.
- Cupples, C.M. et al. (2009) STR profiles from DNA samples with "undetected" or low Quantifiler™ results. J. Forensic Sci. 54, 103–7.
- Walsh, P.S., Erlich, H.A. and Higuchi, R. (1992) Preferential PCR amplification of alleles: Mechanisms and solutions. PCR Meth. Appl. 1, 241–50.
- Findlay, I. et al. (1997) DNA fingerprinting from single cells. Nature 389, 555–6.
- Gill, P. et al. (2000) An investigation of the rigor of interpretation rules for STRs derived from less than 100 pg of DNA. Forensic Sci. Int. 112, 17–40.
- Whitaker, J.P., Cotton, E.A. and Gill, P. (2001) A comparison of the characteristics of profiles produced with the AMPFlSTR® SGM Plus™ multiplex system for both standard and low copy number (LCN) STR DNA analysis. Forensic Sci. Int. 123, 215–23.
- Caragine, T. et al. (2009) Validation of testing and interpretation protocols for low template DNA samples using AmpFlSTR® Identifiler®. Croat. Med. J. 50, 250–67.
- Taberlet, P. et al. (1996) Reliable genotyping of samples with very low DNA quantities using PCR. Nucleic Acid Res. 24, 3189–94.
- Buckleton, J. (2009) Validation issues around DNA typing of low level DNA. Forensic Sci. Int. Genet. 3, 255–60.
How to Cite This Article
Scientific Style and Format, 7th edition, 2006
Butler, J.M. and Hill, C.R. Scientific Issues with Analysis of Low Amounts of DNA. [Internet] 2010. [cited: year, month, date]. Available from: https://www.promega.com/es-es/resources/profiles-in-dna/2010/scientific-issues-with-analysis-of-low-amounts-of-dna/
American Medical Association, Manual of Style, 10th edition, 2007
Butler, J.M. and Hill, C.R. Scientific Issues with Analysis of Low Amounts of DNA. Promega Corporation Web site. https://www.promega.com/es-es/resources/profiles-in-dna/2010/scientific-issues-with-analysis-of-low-amounts-of-dna/ Updated 2010. Accessed Month Day, Year.
Contribution of an article to Profiles in DNA does not constitute an endorsement of Promega products.
Products may be covered by pending or issued patents or may have certain limitations. More information.
All prices and specifications are subject to change without prior notice.
Product claims are subject to change. Please contact Promega Technical Services or access the Promega online catalog for the most up-to-date information on Promega products.