The Advantages to Using Arg-C, Elastase, Thermolysin and Pepsin for Protein Analysis
Sergei Saveliev1, Laurie Engel1, Ethan Strauss1, Richard Jones2 and Mike Rosenblatt1
1Promega Corporation; 2MS BioWorks, LLC.
Abstract
Alternative proteases can improve mass spectrometry protein analysis by using unique digestion conditions or complementary cleavage specificity. In this article, we use four alternative proteases: Arg-C, elastase, thermolysin and pepsin, and show typical applications for these proteases and how their use can improve mass spectrometry analysis. We demonstrate the advantages of these proteases using various model systems, including a yeast total protein extract, a post-translation modification-rich human histone H4, phosphorylase B and bacteriorhodopsin.
Introduction
Trypsin is the most widely used protease, cleaving proteins with high specificity and generating peptides 7–20 amino acids long with a strong C-terminal charge (1) , ideal for mass spectrometry analysis. However, trypsin has certain limitations. Tightly folded proteins resist trypsin digestion, and inadequate distribution of trypsin cleavage sites in certain proteins or protein domains generates peptides that are too long or too short for mass spectrometry analysis. Membrane proteins often exhibit both resistance to trypsin and few trypsin cleavage sites, requiring alternative approaches when preparing for mass spectrometry (2) (3) . Post-translational modifications (PTMs) present yet another challenge because glycans often limit trypsin access to cleavage sites whereas acetylation or di- and trimethylation of lysine and arginine residues make them resistant to trypsin digestion (4) (5) (6).
We provide several alternative proteases that can be used when trypsin is not informative. Lys-C protease is active under denaturing conditions, offering the means to overcome proteolytic resistance of tightly folded proteins. Chymotrypsin preferentially cleaves at aromatic and other hydrophobic residues and, therefore, can digest hydrophobic proteins. Asp-N and Glu-C proteases add flexibility when choosing protein cleavage sites, providing a solution when trypsin does not generate peptides within the optimal size range or PTMs interfere with trypsin proteolysis.
However, mass spectrometry analysis of proteins digested using Lys-C, Asp-N, Glu-C, chymotrypsin and trypsin rarely produce complete protein coverage. Incomplete sequence coverage decreases the number of PTMs available for analysis and diminishes the ability to distinguish between proteins with a high degree of sequence similarity. Here we show that the proteases Arg-C, elastase, thermolysin and pepsin address these issues by increasing protein sequence coverage or digesting under alternative conditions such as higher temperature or lower pH. We demonstrate the advantages of these proteases using various model proteins or protein mixtures, including a yeast total protein extract, a PTM-rich human histone H4, phosphorylase B and bacteriorhodopsin.
The Arg-C Advantage
Arg-C (clostripain), Sequencing Grade (Cat.# V1881), is a specific endoproteinase isolated from the soil bacterium Clostridium histolyticum. It preferentially cleaves at the C-terminal side of arginine (R) residues. It also cleaves at lysine (K) residues although less efficiently. We evaluated Arg-C for protein analysis in two different experiments. In the first experiment, we studied the use of Arg-C for proteomic analysis. Yeast provides an excellent model proteome because its genome is well annotated. Yeast extract was digested in two parallel reactions, using trypsin in the first reaction and Arg-C in the second, using a conventional protocol consistent with LC-MS/MS analysis (see legend for Figure 1). As expected the trypsin digestion resulted in a high number of peptide and protein identifications (Figure 1). However, many peptides remained elusive. The parallel Arg-C digestion complemented the trypsin digestion by recovering an additional 2,653 peptides and providing a 37.4% increase in the number of identified peptides. Digesting with Arg-C also resulted in an increase in the number of identified proteins. In fact, 138 new proteins were identified in Arg-C digest compared to the parallel trypsin digest, offering a 13.4% increase in the overall number of identified proteins.
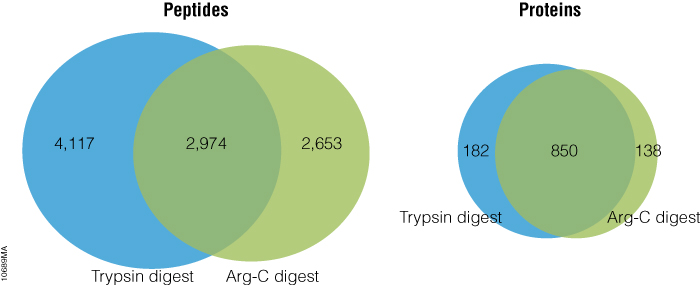
Figure 1. Venn diagrams of the peptides and proteins identified in yeast protein extract digested with trypsin and Arg-C. Yeast total protein extract was reduced, alkylated and digested overnight at 37°C with trypsin or Arg-C at 50:1 protein:protease ratio. Each digest was performed in duplicate. The digests were analyzed with 2 hour gradients by nano LC-MS/MS with a NanoAcquity HPLC system (Waters) interfaced with a LTQ Orbitrap Velos Mass Spectrometer (ThermoFisher). Nonredundant identified peptides and proteins from duplicate digestion reactions with either protease were pooled. The data show that parallel digestion with trypsin and Arg-C increased number of identified peptides and proteins for by 37.4% and 13.4%, respectively, as compared to those identified in the digests with trypsin only.
This experiment also demonstrated that Arg-C efficiently cleaved arginine sites when followed by proline (P). In fact, most RP sites were cleaved in the digests (Table 1). KP sites were also cleaved, although with lower efficiency. Trypsin does not cleave at arginine and lysine residues if they are followed by a proline residue. This difference is important because every twentieth arginine or lysine is followed by proline.
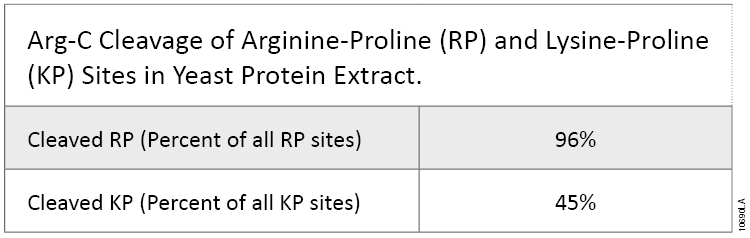
Table 1. Arg-C Cleavage of Arginine-Proline (RP) and Lysine-Proline (KP) Sites in Yeast Protein Extract.
In a second experiment, we tested the ability of Arg-C to analyze individual proteins, selecting human histone H4 as a model protein. Like other histones, this protein is heavily modified by PTMs that alter histone structure and regulate interaction with transcription factors. As a result, histone PTMs are implicated in gene regulation and associated with multiple disorders (7).
Technical challenges, however, impede histone PTM analysis. Histone PTMs are complex and some, such as acetylation and methylation, prevent trypsin digestion, as shown by our data. In our experiment, trypsin digestion of histone H4 identified several PTMs (Figure 2). However, certain PTMs were missing. By digesting histone H4 with Arg-C, we were able to identify the missing PTMs including mono-, dimethylated and acetylated lysine and arginine residues. We speculate that the PTMs in human histone H4, which modified arginine and lysine residues, rendered trypsin unsuitable for preparing the corresponding histone regions for mass spectrometry. The problem was rectified by replacing trypsin with Arg-C.
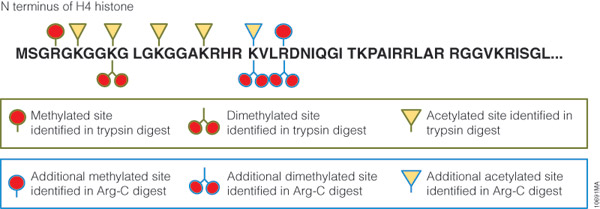
Figure 2. Histone H4 post-translational modifications identified in trypsin and Arg-C digests. Human histone H4 was digested at 37°C for 18 hours with trypsin or Arg-C at 20:1 protein:protease ratio. The digests were analyzed with 1 hour gradients by nano LC-MS/MS with a nanoACQUITY UltraPerformance LC® (UPLC®) System (Waters) interfaced with a LTQ Orbitrap Velos Mass Spectrometer (ThermoFisher).
The Elastase, Thermolysin and Pepsin Advantage
Elastase (Cat.# V1891), thermolysin and pepsin (Cat.# V1959) are nonspecific proteases with a preference for hydrophobic residues. Elastase is isolated from porcine pancreas, thermolysin from the thermophilic bacterium Bacillus thermoproteolyticus rokko and pepsin from porcine stomach. These proteases are relatively small, 26–36kDa. Adapting these proteases for proteomics applications has started relatively recently and is still emerging. With the exception of pepsin, which is extensively used in structural protein studies (8) , these proteases are largely unknown to mass spectrometry users. Nonspecific proteases generate complex peptide pools, which complicates their use for mass spectrometry. Although such complexity might represent a technical challenge in analyzing complex protein mixtures (i.e., cell protein extracts), the peptide pool is manageable for single proteins or simple protein mixtures. The following experiments demonstrate the utility of these proteases for protein mass spectrometry analysis.
Elastase
We used phosphorylase B to demonstrate the benefit of using elastase for protein analysis. A control digestion with Arg-C was used to benchmark elastase performance. Arg-C digestion generated 60% sequence coverage of phosphorylase B (Figure 3). A similar level of phosphorylase B protein coverage was observed for trypsin digestion (data not shown). Elastase was found to significantly improve the protein coverage (Figure 3). The combined sequence coverage for phosphorylase B using Arg-C and elastase approached 90%, demonstrating the advantage of using elastase for protein analysis.

Figure 3. Phosphorylase B sequence coverage obtained in Arg-C and elastase digests. Phosphorylase B was reduced, alkylated and digested overnight with Arg-C at 20:1 protein:protease ratio or for 3 hours with elastase at 50:1 protein:protease ratio. Both reactions were incubated at 37°C. The digests were analyzed with 1 hour gradients by nano LC-MS/MS with a nanoACQUITY UltraPerformance LC® (UPLC®) System (Waters) interfaced with a LTQ Orbitrap Velos Mass Spectrometer (ThermoFisher).
Thermolysin and pepsin
Thermolysin and pepsin are distinct from other proteases because they tolerate extreme conditions: high temperatures and low pH, respectively (9) (10) . These properties make thermolysin and pepsin ideal proteases for the digestion of proteolytically resistant, tightly folded proteins. High temperatures and low pH can denature proteins, allowing thermolysin and pepsin to cleave previously inaccessible sites.
The benefit of using thermolysin and pepsin for protein digestion was demonstrated with bacteriorhodopsin, a bacterial membrane protein containing seven transmembrane domains. Proteolysis of this protein is problematic due to its extreme hydrophobicity and tight conformation (11) . The low number of arginine and lysine residues adds to the digestion challenge. Due to the combination of the above factors, trypsin digestion of bacteriorhodopsin gave low sequence coverage (8.4%) in our study (Figure 4). In contrast, digestion with thermolysin produced high coverage. Heating the reaction to 75°C unfolded bacteriorhodopsin and digesting with the heat-tolerant protease thermolysin increased protein coverage to 61% (Figure 4). Alternatively, digestion with pepsin used low pH for protein denaturation rather than heat. Pepsin digested bacteriorhodopsin more efficiently than thermolysin, providing 86% sequence coverage (Figure 4).
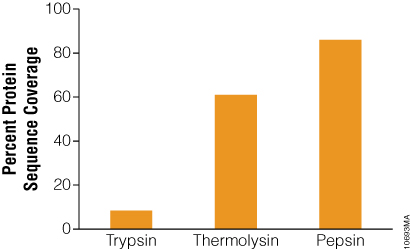
Figure 4. Bacteriorhodopsin sequence coverage obtained from trypsin, thermolysin and pepsin digests. Bacteriorhodopsin was digested with trypsin overnight at 37°C or thermolysin for 2 hours at 75°C at 50:1 protein:protease ratio. Digestion with pepsin was performed in 10mM HCl (pH 2) at 37°C for 3 hours at 20:1 protein:protease ratio. The digests were analyzed with 1 hour gradients by nano LC-MS/MS with a nanoACQUITY UltraPerformance LC® (UPLC®) System (Waters) interfaced with a LTQ Orbitrap Velos Mass Spectrometer (ThermoFisher).
Choosing between thermolysin and pepsin for digestion depends on experimental needs and properties of analyzed proteins. Protein digestion with thermolysin is rapid because the reaction occurs at a higher temperature. However, when high temperature precipitates proteins, pepsin is a viable alternative.
Summary
The proteases also offer flexibility in mass spectrometry protein sample preparation, which can be exploited for specialized applications.
Acknowledgment: We are grateful to Prof. Yali Dou for providing human histone H4 and the MS BioWorks, LLC team for excellent mass spectrometry service.
Arg-C, elastase, thermolysin and pepsin are valuable additions to the protease portfolio. These proteases improve proteomic analysis by increasing the number of peptide and protein identifications in a complex protein mixture and facilitate analysis of individual proteins by allowing more comprehensive PTM mapping and increased protein coverage.
References
- Tran, B.Q. et al. (2011) Addressing trypsin bias in large scale (phospho)proteome analysis by size exclusion chromatography and secondary digestion of large post-trypsin peptides. J. Proteome Res. 10, 800–11.
- Hedin, L.E., Illergård, K. and Elofsson, A. (2011) An introduction to membrane proteins. J. Proteome Res. 10, 3324–31.
- Fischer, F. and Poetsch, A. (2006) Protein cleavage strategies for an improved analysis of the membrane proteome. Proteome Sci. 4, 2–14.
- Lee, J.Y. et al. (2011) Targeted mass spectrometric approach for biomarker discovery and validation with nonglycosylated tryptic peptides from N-linked glycoproteins in human plasma. Mol. Cell. Proteomics 10, 1–12.
- Smith, C.M. (2005) Quantification of acetylation at proximal lysine residues using labeling and tandem mass spectrometry. Methods 36, 395–403.
- Botting, C.H. (2010) Extensive lysine methylation in hyperthermophilic crenarchaea: Potential implications for protein stability and recombinant enzymes. Archaea 2010, 106341.
- Sadri-Vakili, G. and Cha, J.H. (2006) Mechanisms of disease: Histone modifications in Huntington's disease. Nature Clin. Practice Neur. 2, 330–8.
- Wu, Y., Kaveti, S. and Engen, J.R. (2006) Extensive deuterium back-exchange in certain immobilized pepsin columns used for H/D exchange mass spectrometry. Anal. Chem. 78, 1719–23.
- Endo, S. (1962) Studies on protease produced by thermophilic bacteria. J. Ferment. Technol. 40, 346–53.
- Herriot, R.M. (1962) Pepsinogen and pepsin. J. Gen. Phys. 45, 57–76.
- Paul, C. and Rosenbusch, J.P. (1985) Folding patterns of porin and bacteriorhodopsin. EMBO J. 4, 1593–7.